| DISFUNCIÓN
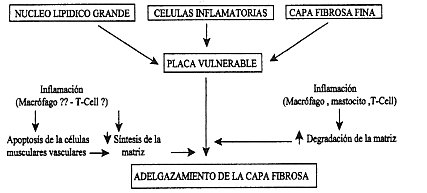
ENDOTELIAL: IMPACTO EN LA ENFERMEDAD ARTERIAL CORONARIA Aun cuando la capa fibrosa varía mucho en su condensación, muchas veces hay poca consistencia en sus regiones de soporte, que es donde más frecuentemente ocurre la ruptura. Las capas de fibras rotas tienden a tener menos células musculares lisas con matriz sintetizada, un más bajo contenido de colágeno y glicosaminoglicano, y un mayor grado de infiltración en células inflamatorias (macrófagos, T-linfocitos, y mastocitos), que las que hacen intactas a las placas. Además, la capa fibrosa infiltrada por macrófagos tiene un umbral mecánico mas bajo para la ruptura cuando es testeado in vitro. Respuesta Inflamatoria en la Placa Varios estudios histomorfométricos han demostrado que las placas rotas contienen una activa infiltración inflamatoria, comúnmente ubicada en la capa fibrosa y rodeando el núcleo lipídico, con una preferencial concentración en los soportes de la placa o debajo de la delgada o rota capa fibrosa externa. Los macrófagos monocitoderivados, frecuentemente sufren marcas de contacto de activación, siendo los más abundantes componentes de la respuesta inflamatoria, pero los linfocitos T activados y los mastocitos desgranulados activados también se encuentran en grandes cantidades en las placas rotas. Evidencia de Desregulación de la Matriz en la Ruptura de Placas: Los Roles de la Inflamación, las Metaloproteinasas, y la Apoptosis La ruptura de las placas resulta de la delgadez, debilidad, y eventual ruptura o fisura de la capa fibrosa. Puesto que la resistencia de tensión de la capa fibrosa está determinada por sus componentes de la matriz, especialmente por el contenido de colágeno, la pérdida del colágeno de la matriz representa un paso crítico que propicia la ruptura de la placa. La pérdida de colágeno puede resultar por la excesiva degradación o la reducida síntesis de la matriz (por ejemplo desrregulación de la matriz)36. Varios estudios recientes tienen demostrado que los macrófagos y, en una menor extensión, las células musculares lisas derivadas en placas aterosecleróticas, producen una familia de metaloproteinasas de degradación de la matriz (MMPs), que son virtualmente capaces de degradar todos los componentes de la matriz extracelular. Factores que están involucrados en la estimulación de la producción o la activación de las MMPs en las placas ateroescleróticas, no está plenamente entendido, pero puede incluir la interacción de los macrófagos con los sustratos, ingestión lipídica por los macrófagos, lipoproteínas oxidativamente modificadas, stress oxidativo, stress mecánico, linfocitos T activados, citoquinas, agentes infecciosos como la clamydia, y otros. Experimentos in vitro y estudios zimográficos han demostrado un neto incremento en la actividad de degradación de la matriz en regiones propensas a ruptura de la placa ateroesclerótica. Algunos antecedentes también muestran reducción del contenido de colágeno y glicosaminoglicanos y una menor cantidad de células musculares lisas en la ruptura de la capa fibrosa. El mecanismo preciso que contribuye a la pérdida de las células musculares lisas no queda claro, pero puede incluir la inhibición de las células musculares lisas debido a la apoptosis o necrosis, la duplicación o incremento de la mortandad de las células musculares lisas. Estudios in vitro de nuestros laboratorios han demostrado que los macrófagos en el cultivo de la célula así como en la placa ateroesclerótica generan tenascina-C, una novedosa proteína de la matriz antiadhesiva que puede, in vitro, inducir apoptosis en las células vasculares musculares lisas (datos no publicados). Estas observaciones aumentan la posibilidad de que las células inflamatorias (por ejemplo los macrófagos) puedan no solamente contribuir a la ruptura de la placa (por degradación de la matriz mediada por MMP) sino también a la mortandad de la célula muscular lisa y, en consecuencia, una reducida síntesis matricial. Causas Mecánicas y Hemodinámicas de la Ruptura de la Placa Una variedad de fuerzas mecánicas y hemodinámicas locales someten a las placas coronarias a un constante stress que puede "causar" la ruptura de placas inestables o vulnerables, particularmente en el punto de su máxima debilidad, la región de soporte de la capa fibrosa. Por ejemplo, placas que son sometidas repetitivamente a curvatura, compresión, estiramiento, deslizamiento, o presión constante, pueden llegar a debilitarse y eventualmente sufrir una ruptura espontánea debido a la "fatiga de la capa", un fenómeno análogo a la fatiga del metal. De acuerdo con la ley de Laplace, el esfuerzo de tensión o de tracción se correlaciona positivamente tanto con la presión sanguínea como con el diámetro luminal (figura 6).
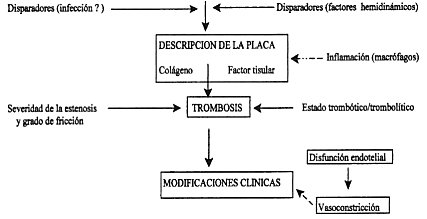
Consecuentemente, las placas que están solo mediana o moderadamente estenóticas y poseen un lumen residual agrandado pueden estar sujetas a un mayor stress circunferencial, haciéndose potencialmente más vulnerables a la ruptura que las placas severamente estenóticas. El máximo stress, usualmente, se desarrolla en el punto donde la capa fibrosa es más delgada, pero puede ocurrir en otros sitios que hubiesen tenido debilidad por la infiltración focal de macrófagos. Las placas también pueden ser sometidas separadamente a esfuerzos constantes, como un resultado de la mecánica del stress que ocurre cuando los tejidos con diferentes propiedades pasan de un estado a otro causando un desgarro en la placa conocido como falla por deslizamiento. El oscilar del pulso puede producir cambios cíclicos en la dimensión y forma del lumen, resultando en la curvatura y deformación de las placas, particularmente aquellas con un gran núcleo ateromatoso. La curvatura cíclica es más probable de ocurrir en la unión entre la placa excéntrica dura y la pared luminal menos enferma (por ejemplo el "borde" de la placa), causando en última instancia debilidad a este sitio, que conduce a la ruptura espontánea. Sin embargo, una curvatura repentina y más intensa puede también causar la ruptura de la capa debilitada. Vasoespasmos, el colapso pasivo de una estenosis, o el flujo de sangre por vasa vasorum, pueden comprimir la placa causando un incremento en la presión intra-placa. Hay una pequeńa evidencia de que sólo los vasoespasmos precipitan la ruptura de placas o la trombosis luminal, o que las sangrías causadas por la ruptura de diminutos capilares de la vasa vasorum podrían incrementar la presión intra-placa, lo suficiente como para inducir a la ruptura de la misma. Ruptura de la Plaqueta y Trombosis La ruptura de la placa puede conducir a la trombosis coronaria; que esto ocurra, depende de la trombogenicidad de los componentes expuestos de la placa, la severidad de la estenosis local, y el equilibrio trombótico-trombolítico sistémico. Los principales componentes trombogénicos de la placa son el colágeno y el núcleo lipídico, aunque el núcleo lipídico puede estar más trombogénico que el colágeno de la matriz. La mayor trombogenicidad del núcleo lipídico puede deberse a su alto contenido de factor tisular catalíticamente activo, una glicoproteína transmembrana, procoagulante, producida mayormente por macrófagos en la placa aterosclerótica. Cuando son expuestos al torrente sanguíneo, los factores tisulares interactúan con el factor VIIa y forman un complejo que activa el factor X. Activado el factor Xa se inicia la cascada por clivaje de protrombina a trombina la cual, a su vez, causa la coagulación y activación de la plaqueta que resulta en formación del trombo. Diversos factores extrínsecos afectan la respuesta trombótica a la ruptura de la placa. Esto incluye:
Los niveles elevados de fibrinógeno, el incremento de la actividad procoagulante mediana del factor VII, la agregación plaquetaria mejorada, y la fibrinólisis endógena deprimida, están todas asociados con un aumentado riesgo de eventos vasculares aterotrombóticos. La disfunción endotelial puede también contribuir a la ruptura de la placa. El endotelio normal es crítico para la regulación de muchas funciones vasculares, incluyendo la regulación del tono vascular por liberación de vasodilatadores (por ejemplo óxido nítrico) y vasoconstrictores (por ejemplo endotelina) y manteniendo un balance entre trombosis y trombólisis por liberación de agentes antitrombóticos (por ejemplo óxido nítrico, proteína C, ecto-ADPasa y protrombóticos (por ejemplo factor tisular, endotelina), sustancias tan buenas como los profibrinolíticos (por ejemplo activador de tejido plasminógeno) y antifibrinolíticos (por ejemplo inhibidor-1 activador de plasminógeno). La disfunción endotelial asociada con ateroesclerosis y la presencia de factores de riesgo por ateroesclerosis, incrementan el potencial por excesiva y paradójica vasoconstricción, expresión de adhesión de moléculas que restablecen monocitos y otras células inflamatorias dentro de la pared arterial, y la promoción de un estado protrombótico y antifibrinolítico. Por lo tanto, las anormalidades en la función endotelial también pueden jugar un rol importante en la trombosis con la subsiguiente ruptura de placa. Manifestaciones Clínicas de la Ruptura de Plaqueta La ruptura de placa no siempre resulta en trombosis y un evento coronario clínico. En efecto, la ruptura de placa es frecuentemente asintomática, y el crecimiento rápido de la placa, frecuentemente es clínicamente silencioso. Estudios de autopsia han demostrado que el 9% de las personas ostensiblemente sanas y hasta 22% de pacientes diabéticos e hipertensos tienen en forma asintomática, ruptura de placas en sus arterias coronarias. No obstante, la ruptura de placas coronarias con y sin trombos es común en pacientes que mueren de enfermedad isquémica coronaria. Es de particular notoriedad aquel paciente que muere como resultado de una enfermedad coronaria aguda teniendo un promedio de más de dos placas rotas cada uno; poco menos de la mitad de éstos, están asociados con trombosis luminal suficiente para causar una obstrucción crítica del flujo sanguíneo. Las manifestaciones clínicas de la ruptura de placa y trombosis varían de acuerdo al grado, localización y duración de la isquemia de miocardio. Por ejemplo, una trombosis oclusiva relativamente estable, es probable que cause un infarto agudo de miocardio a onda Q, mientras que una trombosis no oclusiva (o transitoriamente oclusiva), es más probable que cause una angina inestable, o un infarto de miocardio de onda no Q. La oclusión coronaria total o subtotal puede estar asociada con la muerte cardíaca repentina. Sin embargo, la oclusión coronaria no necesariamente progresa al IAM, si hay circulación colateral adecuada en el momento de la oclusión. No obstante, la ruptura de placa seguida por variables grados de hemorragias dentro de la placa y la trombosis luminal, pueden acelerar más el crecimiento de la placa y la progresión de estenosis, explicando así la progresión repentina, no lineal e impredecible de ateroesclerosis coronaria, al síndrome coronario agudo Pacientes con enfermedad cardíaca coronaria demostraron tener una incidencia serológica relativamente alta de reciente infección por Clamydia pneumoniae37. Conectada con el descubrimiento en las placas ateroscleróticas del antígeno Clamydia, esta observación sugiere la intrigante hipótesis de que una activa inflamación o una respuesta inmune impulsada por infección, puede ayudar a la iniciada aterogénesis y posiblemente también a la ruptura de la placa y a la trombosis. Estabilización de la Placa y Prevención del Síndrome Coronario Agudo En los diversos ańos transcurridos, un número de ensayos angiográficos en serie que evaluaron la eficacia de la disminución de lípidos, o las modificaciones del estilo de vida, tienen demostrada una mayor reducción en la incidencia de los eventos clínicos aterotrombóticos (por ejemplo síndromes coronarios agudos y accidentes cerebro vasculares). Estos podrían esperarse, fundamentados sobre los relativamente menores cambios observados, en la severidad de la estenosis coronaria. Esta paradoja clínica angiográfica condujo al concepto de que la modificación del factor de riesgo, especialmente la disminución de los lípidos, puede reducir los eventos clínicos disminuyéndola incidencia de ruptura de placa y trombosis mediante cambios en la biología de la placa, más bien que por efectos sobre el tamańo o volumen total de la placa o por la severidad de la estenosis. Esta así llamada "estabilización de placa" puede ser alcanzada disminuyendo el contenido lípídico de la placa, cambiando la magnitud de contenido y actividad de la célula inflamatoria dentro de la placa, mejorando la función endotelial, o cambiando en equilibrio trombótico-trombolítico en la circulación. De este manera, la estabilización de la placa puede reducir la vulnerabilidad a la ruptura de las placas o la subsiguiente respuesta trombótica a la ruptura. Este nuevo paradigma terapéutico se aproxima a modernas investigaciones para reducir las consecuencias adversas de la ateroesclerosis, influenciando la biología de la placa más bien que su tamańo o la severidad de la estenosis. Las intervenciones para la disminución o eliminación de lípidos que pueden promover a la estabilización de la placa (incluyendo la improvisada función endotelial y el reducido estado protrombótico) incluyen inhibidores de la enzima convertidora de angiotensina, ? bloqueantes, estrógenos, antioxidantes y el hecho de no fumar cigarrillos. |
||||
|
|
|||