| DISFUNCIÓN
ENDOTELIAL: IMPACTO EN LA ENFERMEDAD ARTERIAL CORONARIA Función Endotelial, Fibrinólisis, e inhibición de la Enzima Convertidora de Angiotensina El Sistema Fibrinolítico El activador de plasminógeno, o sistema fibrinolítico, constituye uno de los primeros mecanismos endógenos para la prevención de la trombosis intravascular, la cual es importante en la patogénesis del infarto de miocardio (IAM) y otros síndromes coronarios agudos. La fibrinólisis depende del balance entre los activadores de plasminógeno (la uroquinasa y el activador del tejido tipo plasminógeno [TPA]) y el inhibidor del activador de plasminógeno tipo 1 (PAI-1), el mayor inhibidor fisiológico de la uroquinasa y la TPA en plasma. Este balance es mantenido a través de los procesos que parecen estar mayormente mediados con el endotelio. Los activadores plasminógenos convierten al plasminógeno en la enzima activa, plasmina, la cual es una proteinasa que lisa los coágulos de fibrina. Un importante mecanismo para regular la generación de plasmina implica la formación de complejos entre el PAI-1 y los activadores de plasminógeno, los cuales previenen la conversión del plasminógeno en plasmina30.31. Dado que ambos, el TPA y el PAI-1 son sintetizados primariamente por las células endoteliales (y las células lisas de la musculatura), se considera que el endotelio juega un rol prominente en la manutención del balance fibrinolítico vascular. El disbalance de las proteínas fibrinolíticas puede también tener consecuencias patogénicas dentro de la pared vascular. En el tejido vascular, la plasmina activa la matriz de las metaloproteinasas, las cuales son cruciales para la remodelación del subsiguiente dańo vascular, la continua degradación de colágeno y otras glicoproteínas que se acumulan en las plaquetas. La especulación de que el endotelio sirve como un enlace entre el sistema fibrinolítico y el sistema renina-angiotensina (RAS) ha sido sostenido por una cantidad de descubrimientos experimentales y clínicos. Se había demostrado que la angiotensina II, era capaz de unir las células endoteliales y simular la producción de dosis dependientes de PAI-1 en el cultivo de células vasculares musculares lisas de la rata, el cultivo de las células aórticas bovinas y las células endoteliales humanas32. Un ligante local de angiotensina recientemente identificado, el receptor de angiotensina IV (AT4), parece ser el receptor en las células endoteliales que se interpone a la expresión PAI-1 en respuesta a la angiotensina33, dando cuenta de la observación que los receptores inhibidores de angiotensina del tipo 1 y tipo 2 dejan de prevenir la producción endotelial de PAI-1. El incremento en la expresión PAI-1 en células cultivadas está condicionado a la conversión del octapéptido de angiotensina al hexapéptido de angiotensina IV, la cual es consumada vía los efectos de aminopéptidos específicos que están localizados en la superficie vascular. Por lo tanto, las respuestas a la angiotensina pueden ser mediadas por un receptor endotelial específico para angiotensina IV, que es el receptor AT4. Conclusión Hay acumulación de evidencia que el RAS interactúa con el sistema fibrinolítico a nivel del endotelio. En la fibrinólisis, tanto la angiotensina II como la ACE, pueden ser considerados protrombóticos: la angiotensina II porque lo induce a la expresión PAI-1, y la ACE porque media la formación de angiotensina II y la degradación de bradiquinina. Los niveles aumentados de PAI-1 en humanos, están asociados con un incremento del riesgo de eventos trombóticos. En modelos experimentales, la inhibición de la ACE esta asociada con las reducciones en la expresión PAI-1 tanto en células cultivadas como en tejido. Estos cambios beneficiosos en las variables fibrinolíticas, pueden ser atribuidos a los efectos duales de la ACE de inhibición de formación de la angiotensina II (y, por consiguiente, limitando la producción de PAI-1) y bloqueando la degradación de la bradiquinina (y por eso, acrecentando la producción de TPA de la bradiquinina). Estos mecanismos pueden contribuir a los efectos de protección vascular de los inhibidores de la ACE. Ruptura de Plaquetas y Trombosis Coronaria: Nuevo Discernimiento Dentro de la Patogénesis y la PrevenciónIntroducción Los resultados de la ateroesclerosis coronaria en un espectro de desórdenes clínicos, se extienden desde la ateroesclerosis asintomática y angina estable, hasta un síndrome coronario agudo, tal como una angina inestable, infarto agudo de miocardio (IAM), y muerte cardíaca súbita. Está estimado que del 30 al 40% de los eventos coronarios agudos ocurren sin cuidados previos en personas que ignoran que tienen enfermedad cardíaca isquémica. Por consiguiente, la prevención de los síndromes agudos coronarios es el mayor enfoque en la medicina cardiovascular. Un análisis profundo de la base patofisiológica para los síndromes coronarios agudos puede conducir a una estrategia preventiva más efectiva. Una de ellas es el concepto de estabilización de plaqueta disminuyendo la vulnerabilidad a la ruptura o fisura de la placa ateroesclerótica. Hasta hace poco, se había pensado que el principal mecanismo patofisiológico al cual la ateroesclerosis contribuye a varios síndromes coronarios, había sido una lenta y progresiva obstrucción luminal por la placa ateroesclerótica que eventualmente resultaba en una disminución del flujo sanguíneo coronario y en la subsiguiente isquemia del miocardio. De cualquier modo, ahora está generalmente aceptado que la ateroesclerosis coronaria progresa de forma no lineal, sino frecuentemente de manera abrupta y con rápida progresión de las lesiones coronarias, incluyendo el repentino desarrollo de la total o casi total oclusión; esto es debido mayormente a la trombosis que complica la ateroesclerosis. Diversas autopsias, estudios angiográficos y angioscópicos han demostrado que las manifestaciones más letales de los síndromes de ateroesclerosis coronaria inducida aguda resultaba en una disminución del flujo sanguíneo coronario y en la subsiguiente isquemia del miocardio de una trombosis que aparece, tanto en los sitios de ruptura de plaquetas (60–80% de las veces), como sobre las áreas de erosiones endoteliales superficiales. Aún cuando se pensó que las lesiones severamente estenóticas son más probables, fuera del tiempo estipulado, para avanzar hacia la oclusión total, es evidente ahora una paradoja angiográfica clínica: 60 al 70% de los síndromes coronarios agudos se producen por la evolución de lesiones coronarias que fueron juzgadas de tener solo leve a moderada estenosis (no limitando el flujo). Esta paradoja puede ser explicada por el hecho de que son más las lesiones suavemente que las severamente estenóticas, con un factor de 5 a 10. También es concebible que la rápida transición de moderada estenosis a una oclusión total es más probable que resulte en un evento clínico, porque la colateral protección que normalmente se desarrolla fuera del tiempo estipulado con una lesión severamente estenótica, no puede tener una chance de desarrollo (figura 5).
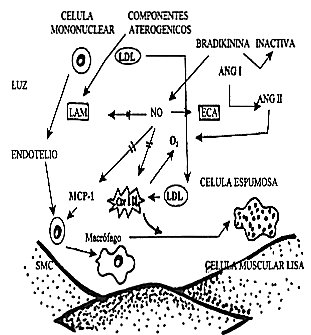
FIG.5: Modificado del Clín. Cardiol. (Suplem.II) Noviembre de 1997. Esquema de los procesos celulares que llevan a la formación de las células espumosas dentro de la luz del vaso. el óxido nítrico (NO) aparentemente modula la expresión de la célula vascular o las móleculas de adhesión leucocitarias (LAM) y/o las proteínas quimiotácticas de los miocitos. El rol vasoprotector dell óxido nítrico puede ser inhibido por la enzima convertidora de la angiotensina, la cual inactiva a la bradikinina. La angiotensina II (ANG II) parece promover la oxidación de las lipoproteínas de abja densidad (LDL) las cuales son fagocitadas por un macrófago para formar las células espumosas. La ANG II y la LDL oxidadas además, pueden actuar sinérgicamente para incrementar la producción del anión superóxido (O2) en cual inactiva al óxido nitrico (NO) y puede directamente causar la contracción de la célula muscular lisa (SMC) Determinantes de la Ruptura de Placa Composición de la Placa Las características histomorfométricas asociadas con la ruptura de placa incluye lo siguiente:
Las placas ateroescleróticas maduras, están conteniendo dos componentes principales: un núcleo rico en lípidos y una matriz extracelular constituida por colágeno y otras proteínas. Alguna matriz de placa está organizada frecuentemente como pobre en lípidos y una capa fibrosa protectora rica en colágeno, que separa el núcleo del lumen. Cuanto más grande la cantidad de matriz extracelular, más "estable" es la placa. Más del 70% de una placa coronaria estenótica típica, consiste en una matriz extracelular sintetizada por células vasculares musculares lisas y con un contenido primordial de colágeno, elastina, proteoglicanos y glicosaminoglicanos34. En contraposición, el núcleo lipídico es usualmente delicado, hipocelular, vascular y ateromatoso, constituido principalmente por lípidos extracelulares (por ej., colesterol y sus ésteres)35. Se pensó que los núcleos ateromatosos ricos en lípidos eran derivados de la necrosis o apoptosis de los macrófagos ricos en lípidos células espumosas y posiblemente también por lipoproteínas en sangre atrapadas dentro del espacio extracelular subendotelial. La composición lipídica del núcleo ateromatoso determina su consistencia. Un núcleo primariamente compuesto por éster de colesterol es blando, mientras que un núcleo conteniendo colesterol cristalino, es duro. Las placas rotas, tienden frecuentemente a tener un gran núcleo blando rico en lípidos y excéntricamente ubicado, que constituye más del 40% del volumen de la placa. |
||||
|
|
|||