|
|
SOCIEDAD DE MEDICINA INTERNA DE BUENOS AIRES |
|
|
SOCIEDAD DE MEDICINA INTERNA DE BUENOS AIRES |
|
Revista de la Sociedad
de Medicina Interna Neumonías Difusas Intersticio Alveolares Las enfermedades pulmonares intersticiales o
neumopatías difusas intersticio-alveolares (NDIA) constituyen un grupo
heterogéneo de procesos caracterizados por la infiltración celular y
no celular de las estructuras alvéolointersticiales. Estas enfermedades tienen en
común rasgos fisiopatológicos, clínicos y radiológicos. Pueden tener un curso agudo, subagudo o
crónico. Si no se resuelven, espontáneamente o tras el oportuno
tratamiento, suelen conducir a una fibrosis pulmonar difusa, destruyendo
las unidades alvéolocapilares y alterando gravemente el intercambio
gaseoso (1, 2) Los términos «enfermedad pulmonar
intersticial» o «neumopatía intersticial», si bien ampliamente
utilizados por numerosos autores y tratados médicos, son erróneos. En
realidad, estas enfermedades no están limitadas al intersticio
pulmonar, habitualmente afectan también a los espacios alveolares, los
bronquiolos terminales, el tejido conjuntivo peribronquial, los vasos
pulmonares y, en ocasiones, el espacio pleural. De hecho, algunas de las
enfermedades catalogadas como intersticiales son procesos principalmente
intraalveolares, con escaso o casi nulo componente intersticial (1). El número de NDIA es elevado e incluye
aproximadamente unas 140 a 200 entidades distintas. La clasificación
utilizada más a menudo es la que las divide según su etiología (tabla
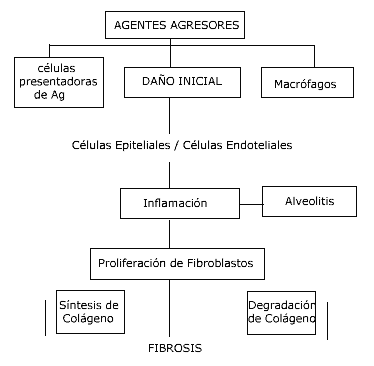
I) (1,2). Patogenia Independientemente de su etiología, la
patogenia de las NDIA es un proceso integrado por dos componentes
básicos: uno inflamatorio, de alveolitis, y otro dinámico, de
reparación y fibrosis. Aunque en general se considera que la alveolitis
acontece primero y la fibrosis después, lo cierto es que ambos
fenómenos suelen ocurrir simultáneamente, si bien no de manera
uniforme. Es decir, es habitual que coexistan zonas de pulmón normal
con áreas de inflamación activa y zonas de fibrosis sin evidencia de
inflamación (1). Los eventos iniciales en la patogénesis de la
fibrosis intersticial son difíciles de analizar, debido a que son
enfermedades crónicas de comienzo insidioso y se diagnostican en etapas
avanzadas. Dependiendo del agente agresor, la magnitud del
dańo y el comportamiento de los mecanismos de defensa, la célula del
parénquima pulmonar puede ser estimulada o dańada, originando necrosis
del endotelio o epitelio y/o estimulación exagerada de las células
inflamatorias o inmunocompetentes (Fig 1) Agentes
Agresores Externos Enfermedades de
Etiología Desconocida La alveolitis en las NDIA se caracteriza por la
acumulación de células inflamatorias e inmunoefectoras en las paredes
alveolares y los espacios aéreos. Esta acumulación se inicia como
respuesta a una lesión tisular ocasionada por agentes que en ocasiones
son conocidos (radiación, polvo orgánicos o inorgánicos,
citostáticos), pero que en otras se desconocen. La infiltración
celular es variada: macrófagos, linfocitos, neutrófilos, eosinófilos
o células plasmáticas. El predominio de una u otra estirpe celular
normalmente depende del tipo de proceso y de su etiología específica,
aunque es posible distinguir dos grandes grupos, uno que presenta un
componente granulomatoso en su alveolitis y otro, en donde existe
predominio de neutrófilos, como en la fibrosis pulmonar idiopática. En el primero (p. ej., la sarcoidosis y las
neumopatías por hipersensibilidad o alveolitis alérgicas
extrínsecas), se cree que el linfocito desempeńa un papel patogénico
primordial, por lo que se designan como alveolitis linfocitarias. En el
segundo grupo, el neutrófilo desempeńa una función patogénica
esencial, considerándose como una alveolitis neutrófila. Cuando entre la alveolitis (fase inflamatoria) y
los mecanismos de reparación y restauración tisular se produce un
desequilibrio, el proceso puede progresar hacia una fibrosis pulmonar
difusa (1). De los dos tipos de alveolitis la más fibrosante y, por lo
tanto, de peor pronóstico es, sin duda, la neutrófila, probablemente
por ser el neutrófilo una célula muy tóxica, capaz de liberar enzimas
proteolíticas y sustancias oxidantes (radicales libres de oxígeno). Denominamos a este heterogéneo grupo de
enfermedades como intersticioalveolares por la participación del
compartimento intraalveolar en su desarrollo. Luego del dańo inicial se
pone en marcha el mecanismo de la inflamación, con proliferación de
neutrófilos que activan a los macrófagos alveolares, liberando
numerosas citoquinas responsables de la estimulación de los
fibroblastos, originando nuevos desequilibrios. La agresión pulmonar
inicial puede provocar lesión en el endotelio capilar y en las células
epiteliales y poner en marcha los mecanismos de la inflamación,
alveolitis. Éstos llevan a una proliferación de fibroblastos con
alteraciones en la síntesis y degradación del colágeno. El mecanismo de progresión hacia la fibrosis
comprende una secuencia de fenómenos aún mal conocidos. Se demostró
que en estadios iniciales existe aumento transitorio del colágeno, con
aumento del colágeno Tipo I sobre el Tipo III, (relación normal I /
III es 2/1). En estadios avanzados se observa una disminución de la
actividad colagenolítica, con predominio del colágeno tipo I. El proceso de fibrosis se manifiesta tanto con
fibrosis intraalveolar como intersticial para luego evolucionar a un
patrón de total remodelamiento que lleva al pulmón al estadio final de
panalización. La fibrosis pulmonar idiopática (FPI), también
denominada alveolitis fibrosante criptogénica por los ingleses (2) fue
descripta por primera vez, en su forma de evolución fulminante, por
Hamman y Rich en 1935. Después de la Sarcoidosis, es la NDIA de causa
desconocida más frecuente, con una incidencia anual aproximada de 3 a 5
casos por cada 100.000 habitantes. En la serie de Coutals y col. se
estima que el diagnóstico más frecuente fue la Fibrosis Pulmonar con
45% (primarias y secundarias), le sigue en frecuencia las Neumoconiosis,
Sarcoidosis y enfermedades del colágeno (3). Puede presentarse a
cualquier edad, aunque la media en el momento del diagnóstico suele
estar entre los 50 y los 60 ańos. Existe un ligero predominio en el
sexo masculino. Representa el prototipo de enfermedad,
generalmente progresiva con una supervivencia entre 2 a 5 ańos de
realizado el diagnóstico. Según sus características histopatológicas (TABLA
2) la neumonitis usual (UIP) y descamativa (DIP) representan,
para algunos autores, etapas tempranas y tardías de la misma
enfermedad, mientras que para otros son procesos patológicos
distintos(1) (3). El hecho es que desde el punto de vista clínico y
evolutivo la DIP tiene mejor pronóstico, con una sobrevida cercana al
100%, mientras que la sobrevida media de la UIP a los 6 ańos es del
10%. TABLA 2
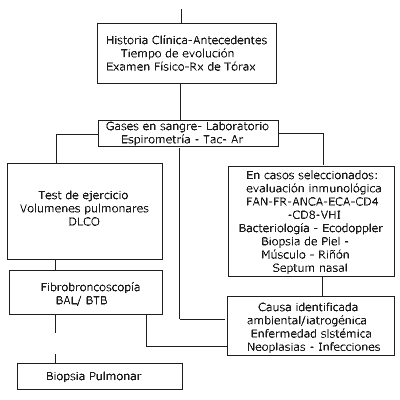
Cuadro clínico y laboratorio En cualquier tipo de evolución el síntoma cardinal es la disnea de esfuerzo progresiva(4). Otros síntomas asociados son la tos seca, hiporexia, astenia y pérdida de peso, el hipocratismo digital y en menor medida la osteoartropatía hipertrófica. La disnea y la tos son los motivos de consulta más frecuentes(4), lo que obliga a incluir estas patologías en el estudio de una disnea o tos persistente no aclaradas. A la auscultación se encuentran estertores crepitantes bibasales (tipo velcro)(4) y en los estadios moderados y avanzados se manifiestan los signos clínicos y electrocardiográficos de hipertensión pulmonar y cor pulmonar. Entre los estudios de laboratorio se puede encontrar aumento de la eritrosedimentación, hipogamaglobulinemia, factor reumatoideo, aumento de los niveles séricos de ECA; los anticuerpos ANCA, antinucleares (FAN) y los complejos inmunes circulantes pueden ser positivos según la etiología específica. Radiología El estudio de los aspectos radiológicos es fundamental en la evaluación de estas enfermedades. El examen radiológico demuestra reducción del volumen pulmonar, con patrones nodulares, micronodulares o reticulares. La radiografía de tórax es normal en el 10% - 15% de los casos y aún en presencia de anormalidades, la Rx de tórax tiene limitada especificidad diagnóstica en pacientes con NDIA (5). La TACAR (TAC de alta resolución) es particularmente útil para detectar enfermedad pulmonar en pacientes sintomáticos o en pacientes con Rx de tórax normal, también nos determina la distribución de las lesiones y nos puede sugerir el grado de actividad(6). En las primeras etapas las imágenes que predominan son las de vidrio esmerilado, compatible con el estadio de alveolitis, por lo tanto el hallazgo de este patrón indica mejor respuesta al tratamiento esteroide y mejor pronóstico y con altas posibilidades de afección reversible. La TACAR ha pasado a ocupar en la última década un lugar muy importante en el estudio de NDIA correlacionando significativamente la severidad de los síntomas y el deterioro funcional(7). Además juega un papel crítico en la evaluación de la actividad de estas enfermedades, para determinar la presencia o ausencia de alteraciones reversibles (agudas o activas) o irreversibles (fibróticas)(8). Cuando la enfermedad evoluciona a estadio terminal es visible el patrón de “Pulmón en Panal” La utilidad de este estudio por lo tanto está relacionada con los parámetros que figuran en tabla 3.
Los estudios isotópicos con Galio 67 permiten detectar la enfermedad en estado activo de alveolitis, siendo importante remarcar que puede ser negativo y no invalidar el diagnóstico ya que en etapas avanzadas la fibrosis reemplaza a la alveolitis. El análisis de depuración del ácido pentaacético de dietilenetriamina (DTPA) marcado con tecnecio 99, se incrementa en los pacientes con dańo pulmonar de cualquier etiología; es por lo tanto demasiado inespecífico para ser de utilidad diagnóstica, pero en los pacientes con aclaramiento del Tc 99 rápido sería indicador de buena respuesta al tratamiento y podría también ser de referencia en qué momento finalizar el tratamiento esteroide. Laboratorio Funcional Respiratorio Desde el punto de vista funcional respiratorio estas entidades presentan un trastorno predominantemente restrictivo caracterizado por una progresiva disminución de los volúmenes y capacidades, aunque algunas enfermedades son excepciones con volúmenes pulmonares normales o levemente elevados; en algunos casos puede coexistir disminución al flujo aéreo. Las pruebas de función respiratoria revelan un patrón restrictivo con reducción de los volúmenes pulmonares y de la distensibilidad. En etapas tempranas de la enfermedad lo más significativo es el descenso de la PO2 durante el ejercicio, siendo ésta una manifestación precoz de la FPI y posteriormente con el avance de la fibrosis se presenta la hipoxia durante el reposo. La afectación del intercambio gaseoso es debida al desequilibrio ventilación / perfusión. Se manifiestan por el descenso de la capacidad de difusión del monóxido de carbono (DLCO). El estudio del intercambio gaseoso también revela desaturación en ejercicio a través del test de caminata de 6 minutos, recordando que el ejercicio ejerce un profundo defecto sobre las pruebas de función respiratoria, en general produce una respiración rápida y superficial con una caída significativa de la PO2 con aumento de la diferencia alvéolo-arterial de oxígeno (A-a O2). Es importante seńalar que las alteraciones observadas en la mecánica respiratoria y en el intercambio de gases son compartidas por la mayoría de las NDIA y por lo tanto no aportan sobre el diagnóstico específico, pero sirven para evaluar la gravedad y controlar la evolución de la enfermedad y la respuesta al tratamiento. Métodos Diagnósticos Todo infiltrado difuso en la Rx de tórax, disnea de causa inexplicada, o incapacidad ventilatoria restrictiva y/o hipoxemia con desaturación en ejercicio, justifican la evaluación para confirmar o descartar la presencia de enfermedad intersticioalveolar. En el lavado broncoalveolar (BAL), se observa un incremento del número de neutrófilos, eosinófilos y linfocitos (CD4-CD8) con diferentes patrones para cada etiología. Puede ser útil en algunas afecciones (hemorragia pulmonar, histiocitosis X, proteinosis alveolar, enfermedades ocupacionales) pero sólo sugestivo en otras. Hay que remarcar que la especificidad es relativa ya que requiere un procesamiento técnico riguroso y en laboratorios entrenados. Es importante seńalar que estos hallazgos celulares se ven modificados por el hábito tabáquico, con aumento del porcentaje de neutrófilos en los pacientes tabaquistas. El método diagnóstico definitivo es la biopsia pulmonar, ya sea por BTB, videotoracoscopía o a cielo abierto. Es la herramienta más precisa, porque no sólo sirve para el diagnóstico, sino que también desempeńa un papel fundamental en la planificación del tratamiento, estadificando la enfermedad y orientando el pronóstico. La biopsia pulmonar a cielo abierto frecuentemente es el elemento que se requiere para establecer el diagnóstico de enfermedad intersticial (9) y en los casos de duda diagnóstica es el estándar de oro. Sus indicaciones están relacionadas con los puntos enumerados en la tabla 4
La rentabilidad en el diagnóstico de la biopsia a cielo abierto ronda el 90%, con bajo porcentaje de morbilidad y mortalidad (10). La BTB es útil en patología con lesiones adyacentes al bronquio, tiene buen rendimiento en sarcoidosis, linfangitis carcinomatosa, infecciones y hemorragia pulmonar. Sus complicaciones incluyen el neumotórax y la hemorragia. El rendimiento de la misma fue evaluado en distintos estudios, Wall y col. verificaron: tejido insuficiente en un 20%, tejido normal 15%, diagnóstico positivo 37.7% (11). Se debe tener en cuenta que la toma es a ciegas, y que las áreas adyacentes al bronquio tienen lesiones fibróticas inespecíficas. La BTB sólo es útil cuando la zona comprometida se sitúa en la zona parahiliar o enfermedades broncocéntricas (sarcoidosis, neumoconiosis) pero en los casos de lesiones basales y subpleurales definitivamente el estudio a realizar es la biopsia pulmonar a cielo abierto o videoasistida (12). Complicaciones En las etapas avanzadas de la enfermedad además del deterioro funcional respiratorio de los pacientes, muchos de ellos sufren distintas complicaciones asociadas a la enfermedad o la terapéutica inmunosupresora. Las causas de morbimortalidad incluyen(13): -Insuficiencia respiratoria y desarrollo de cor pulmonar
Respecto de la evolución, algunas son muy agresivas y ocasionan la muerte en pocos ańos, otras en cambio tienen mejor pronóstico. En cuanto a este hecho hay trabajos que demuestran que el pronóstico depende en muchos aspectos del grado morfológico de fibrosis al momento del diagnóstico, esto nos demuestra la necesidad de hacer un diagnóstico precoz en los casos de neumopatías difusas. Tratamiento de las NDIA Se ha aceptado que el tratamiento de las enfermedades fibrosantes pulmonares tiene como objetivo reducir la progresión de la inflamación hacia fibrosis, o sea que estaría indicado cuando predominan lesiones alveolíticas, pero las decisiones no son tan definitivas cuando nos encontramos frente a cada caso en particular. Sería importante que al actualizar el tema, quedaran en claro ciertas premisas básicas: qué pacientes deben ser tratados, cuándo iniciar el tratamiento, elección del fármaco, cuáles son los parámetros de control de respuesta, duración del tratamiento, etc. Conviene remarcar que hay dos cuestiones invariables que dificultan per se la consideración del tratamiento de la fibrosis pulmonar: 1- La FP es una afección progresiva y letal en un plazo relativamente breve, con una vida media de alrededor del 50% a los 5 ańos del diagnóstico (14), que no se ha modificado con la implementación de distintos esquemas terapéuticos, incluido el trasplante de pulmón (15). 2- No hay estudios con validez técnica que permitan normatizar el tratamiento de FP, ya que no son prospectivos, randomizados, seleccionados al azar, con gran número de casos ni de largo seguimiento (14). Tras estas consideraciones, se puede llegar a consensuar un abordaje terapéutico racional sobre la base de las escasas investigaciones básicas y a los numerosos reportes mencionados de ensayos clínicos. Para establecer el grupo de pacientes con FP que debe recibir tratamiento es importante tener en cuenta aquellos factores que se han visto asociados a mayor progresión de la enfermedad (16) y los que condicionan respuesta favorable al tratamiento (17)(Tabla 5)
Teniendo en cuenta estos factores de preselección: alveolitis predominante + riesgo de evolución a fibrosis + probabilidad de respuesta, se recomienda iniciar el tratamiento lo antes posible, estableciendo parámetros de control de evolución y respuesta al mismo. Para los pacientes que se presentan con fibrosis avanzada, si bien no se proscribe el tratamiento farmacológico, se debe tener presente que las posibilidades de respuesta son mínimas, a la vez que se mantiene el riesgo de efectos adversos. Selección del tratamiento La farmacoterapia por excelencia en FP es con corticoides, y entre éstos, la prednisona. En la tabla 6 se presentan los esquemas terapéuticos empleados, con más detalles sobre la misma y mención de las demás drogas propuestas como alternativa. Cabe mencionar que la mayoría de los autores han evaluado combinaciones de prednisona más un citostático o inmunomodulador y es excepcional que se plantee el uso de estas drogas alternativas como primera elección, salvo, por supuesto, en caso de que haya contraindicaciones absolutas para corticoterapia (18). Duración del tratamiento y evaluación de respuesta al mismo La fibrosis pulmonar idiopática es la que más ha sido evaluada en protocolos terapéuticos. Diversos autores han reportado una respuesta favorable inicial en aproximadamente un tercio de los pacientes tratados (19,20), aunque muchos presentan, luego de esta respuesta inicial favorable, una falta de mejoría a lo largo de los meses sucesivos. La sarcoidosis y la neumonitis por hipersensibilidad en estadio temprano suelen responder bien al tratamiento. La dosis inicial de 1 a 1,5 mg/K/d de prednisona oral se mantiene durante 8 a 12 semanas, momento en que se reevalúa el paciente con estudios de imagen y funcionales. Si se constata estabilidad o mejoría, se disminuye la dosis a 0.5-1 mg/k/d durante 12 semanas y se reevalúa al paciente, si hay estabilidad o mejoría se disminuye a 0,25 mg/k/d. Los parámetros para determinar respuesta favorable al tratamiento son (21):
Si entre los 3 y 6 meses de iniciado el tratamiento se mantiene estabilidad o remisión de la enfermedad, se considera al paciente como respondedor. La duración del tratamiento no está definitivamente establecida. Si hay franca remisión lesional se suspende al cabo de uno o dos ańos, algunos autores proponen mantener dosis de mantenimiento de por vida. Se pueden presentar episodios de reactivación que obligan a reiniciar dosis plenas. Cuando no se constata estabilidad o mejoría, se debe determinar la condición de no respondedor y suspender el tratamiento. La respuesta a esteroides se presenta en alrededor del 15% al 30% de los pacientes (19, 20). No está claro si la mejor sobrevida asociada a mejor respuesta a esteroides es causa o consecuencia y si la presunta respuesta de UIP a esteroides no se trata de casos de NSIP incorrectamente clasificados. La opción de drogas alternativas se evalúa ante falla del tratamiento esteroideo. Debe tenerse en cuenta que la respuesta a los esteroides es precoz (2-3 semanas), pero la respuesta a inmunosupresores es más lenta (no antes de 3-12 meses). Tabla 6
Las alternativas de tratamiento son la ciclofosfamida, azatioprina, ciclosporina o colchicina. La mayor parte de los estudios con estos agentes no presenta marcadas ventajas sobre el tratamiento esteroide. Se debe considerar fallo terapéutico y eventual agregado de un citostático cuando se encuentra: Progresión lesional radiológica (panalización
o signos de hipertensión pulmonar). Insistimos en el concepto preenunciado de que ningún estudio que haya demostrado respuesta favorable a la corticoterapia ha establecido cambios en la sobrevida a 5 o más ańos. Es interesante destacar uno de los estudios de supervivencia más amplio y prolongado, realizado por Izumi y col. de Japón (22), donde controlaron prospectivamente durante 10 ańos 222 pacientes con FPI y encontraron que no hubo diferencias significativas en la sobrevida de los pacientes que habían recibido esteroides respecto de los que no los habían recibido (por el contrario, hubo una mayor sobrevida, no significativa estadísticamente, entre los que no recibieron corticoterapia). Figura 2
Bibliografía 1-Selman Lama, M. Neumopatías Intersticiales Difusas 1 Edición, Ed Médica Panamericana, 1996 ( 17-64) 2-Masson, S.A. Medicina Interna. Barcelona, En Fernández Sánchez y col, Enfermedades Pulmonares Intersticiales Difusas 1997; 1102-1110 3- U.S. Departament of Healt, Education, and welfare. Respiratory diseases task forse. Washington D.C: U.S. Government Printing Office; 1972, 1976. Publication n 76-432 4-Coultas DB, Zumwalt RE E et al. The Epidemiology of Interstitial lung diseases. Am J Respir Crit Care Med 1994; 150:967 5-King TE. Approach to the patient with interstitial lung disease en Texbook of Internal Medicine Kelley, W 3rd ed Lippincott Raven Philadelphia 1997; 1954 6-McLoud, TC, Gaensler, EA~ et al. Normal chest roentgenograms in chronic diffuse infiltrative lung disease. N Engl J Med 1978; 298 :934. 7- Muller, NL. Clinical value of high-resolution CT in chronic diffuse lung disease AJR Am J Roentgenol 1991; 157:l163. 8-Nishimura K et al. Diagnostic of high resolution computed tomography in diffuse infiltrative lung disease Chest 1993; 104:1149 9-Leung AN. Et al Parenchymal opacification in chronic infiltrative lung disease Radiology 1993; 188:209 10- Wagner JD. Stahler, C, et al . Cilnical utility of open lung biopsy for undiagnosed pulmonary infiltrates. Am J Surg 1992; 164:104. 11- Hartman DL, Myiet D, Gaither JG. et al, Comparison of thoracoscopic lung biopsy with open lung biopsy in diffuse interstitial lung disorders. Am Rev Respir Dis 1992; 145: 750. 12- Walt CE, Gaensler EA, Carrington CB, Hayes JA. Comparison of transbronchial and open biopsies in chronic infiitrative lung diseases. Am Reu Respir Dis 1981; 123: 280. 13- Panos RJ, Mortenson RL y col Complications of Pulmonary Fibrosis Am J Med 1990; 88:396. 14 - US Public Health Service.Single and double transplantation. Health Technology Assessment Reports. Washington DC: Agency for Health Care Policy and Research Publications Clearinghouse, 1991. DHHS Publications No PB92-156793. 15 - Selman Lama, M. Neumopatías Intersticiales Difusas 1 Edición, Ed Médica Panamericana, 1996 ( 207-217). 16 - Gay, SE y col. Idiopathic Pulmonary Fibrosis. Predicting response to therapy and survival. AM J Resp Crit Care Med 1998, 157:1063. 17 - Brown, K, King T Jr. Recent advances in Interstitial lung disease. In: 1995 Year Book of Pulmonary Disease, Mosby Year Book, St. Louis, 1995. 18 - Dayton CS y col. Outcome of subjects with idiopathic pulmonary fibrosis who fail corticosteroid therapy. Implications for further studies. Chest 1993, 103:69. 19- Schwartz DA y col. Determinants of survival in idiophathic pulmonary fibrosis. Am J Resp Crit Care Med 1994; 149:450. 20 - Turner Warwick M y col. Cryptogenic fibrosing alveolitis: response to corticosteorid treatment and its effect on survival. Thorax 1980; 35:593. 21- Sullivan EJ and King TE. Treatment of Idiopathic pulmonary fibrosis with corticosteroids. In: UpToDate 6.3, 1998. 22 - Izumi y col. Ten year follow up of 222 patients with idiopathic pulmonary fibrosis. Am Rev Resp Dis 1992; 145: A218. |
||||||||||||||||||||||||
![]()