|
Si bien en 1889 Richard Pfeiffer fue el que comprendió y expuso la presencia de esta sus-tancia bactericida que provocaba la bacteriolisis, resultó ser Paul Ehrlich unos ańos más tarde, el primero que concibió al complemento actuando como toxina bacteriana que dańaba a las células e introdujo entonces el término complemento para remarcar el hecho de que existía en el suero fresco una sustancia lábil al calor que "complementaba" a los receptores específicos a los que denominó "amboceptores". Bordet finalmente en 1896 es quien escribe sobre las propiedades citolíticas del suero1.
Hoy se sabe que el funcionamiento del llamado "sistema del complemento" juega un papel esencial en la defensa del huésped contra los agentes infecciosos y en los procesos inflamatorios2. El mismo es un importante componente del sistema inmunitario innato y adquirido3 pero su activación en forma inapropiada o excesiva está involucrada en numerosas condiciones patológicas.
El sistema se compone de un complejo de más de 30 proteínas que funcionan algunas como enzimas y otras como proteínas de unión. El conjunto de las fracciones del complemento constituyen cuantitativamente el 15 por ciento de las globulinas y su concentración llega a más de 3 gramos por litro de plasma.
Además de estas proteínas plasmáticas de fun-ciones variadas, también están incluidas en la estructura de la cascada, múltiples receptores de membranas celulares que tienen especificidad de unión con distintos fragmentos de la degradación fisiológica de las proteínas del complemento, recep-tores que están presentes sobre todo en las células inflamatorias y en las del sistema inmunológico.
Como sucede en todos los mecanismos en cascada que funcionan en los organismos animales, existen también en este caso al mismo tiempo, mecanismos que previenen la activación autóloga para proteger al huésped de un ataque "accidental" del complemento.
El papel del "sistema del complemento" en la defensa del huésped pudo establecerse también en forma fehaciente, tal como ocurrió con la cascada de la coagulación, a partir de las enfermedades que surgen de las deficiencias genéticas de varios componentes de la misma4.
El papel del complemento en la inflamación y en la injuria de los tejidos resultó evidente a través de la investigación clínica y sobre todo por los descubrimientos acerca de que la patogenia de ciertas enfermedades inflamatorias complemento-dependiente.
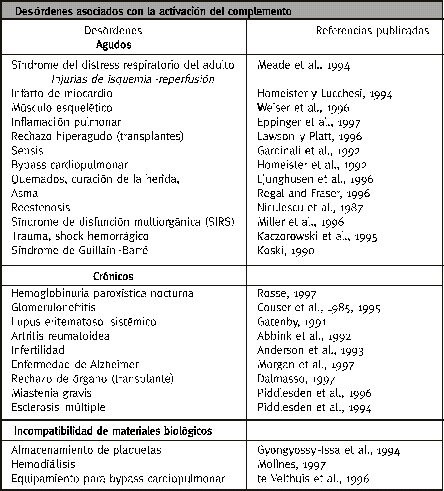
Las múltiples funciones del sistema del complemento pueden compendiarse en tres capítulos5 (tabla 1).

Estas funciones se efectúan por distintos mecanismos tales como:
1) Convirtiendo a las bacterias más susceptibles a la fagocitosis por opsonización
2) Produciendo la lisis directa de algunas bacte-rias o virus
3) Produciendo substancias quimiotácticas
4) Aumentando la permeabilidad vascular
5) Provocando la contracción del músculo liso a través de la degranulación de los mastocitos
El sistema del complemento puede ser activado por tres vías distintas, la denominada "Vía Clásica", la "Vía Alterna" y una vía descripta posteriormente como la "Vía de las
Lectinas". Las tres vías tienen formas de activación distintas que explican luego la posibilidad de diversas
pato-logías a partir de las respectivas fallas (tabla 2).
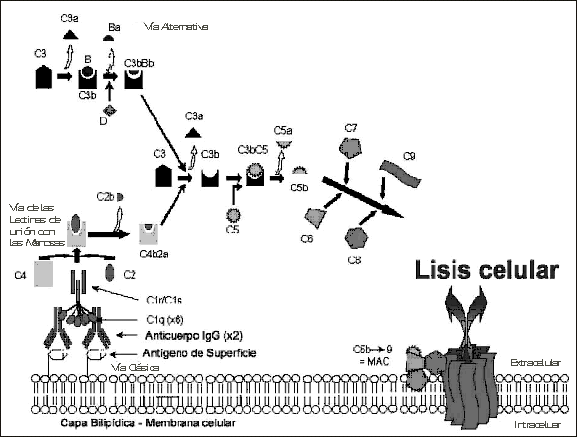
Las tres vías de activación de la cascada del complemento tienen una etapa final común que está constituida por la formación del Complejo de Ataque a la Membrana o MAC y las tres llegan a esta etapa por un hecho también común como es la activación de la fracción C3 (figura 1).
La vía clásica es activada por la unión de una molécula de anticuerpo
(IgG o IgM) con una partícula extrańa siendo por lo tanto anticuerpo-dependiente y funciona mediando en la respuesta específica del anticuerpo. Esta vía es activada por la unión de los antígenos celulares con la molécula de anticuerpo.
El componente C1 está presente en el suero como una proenzima que tiende normalmente a la autoactivación pero está estrictamente controlada por el inhibidor del C1 (C1-in). Esta vía es tan elaborada como la alternativa aunque no posee el mecanismo de la iniciación espontánea ni el mecanismo de amplificación de la regeneración de esta última.
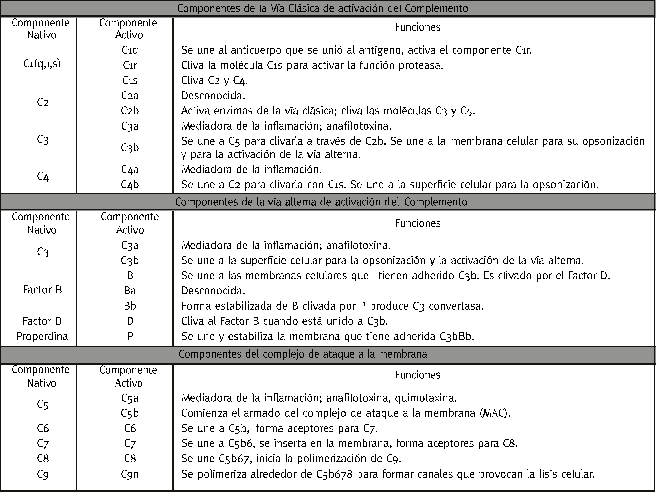
La vía clásica es iniciada por la formación del complejo pentamolecular C1, (este complejo consiste en la unión de una molécula de C1q, dos moléculas de C1r, y dos moléculas de C1s) que se une a anticuerpos que están adheridos previamente a antígenos de la superficie de células bacterianas (ver las distintas fracciones intervinientes en la tabla 3).
Las moléculas C1s del complejo C1 provocan el clivaje de las C4, que se unen también a la superficie de las bacterias, luego cliva a las C2 llevando a la formación del complejo enzimático C4b2a que conforma a la C3 convertasa de esta vía clásica.
La vía alternativa parece ser la de mayor importancia en la defensa del huésped y es activada por los microorganismos invasores sin necesidad de la presencia del anticuerpo siendo precisamente esta vía un componente humoral de la defensa innata que por lo tanto puede actuar sin la presencia de
anticuerpos.

Esta vía es iniciada por la unión de pequeńas cantidades de C3b a los grupos hidroxilo en las proteínas e hidratos de carbono que suelen estar en la superficie de las células. Las pequeńas cantidades de C3b son producidas por un clivaje de bajo grado de la molécula C3 que se produce normalmente en el plasma.
Estas pequeńas cantidades de C3b se unen al factor B, una proteína homóloga a C2 para formar el complejo C3bB. Es el microambiente el que determina que las partículas C3b prefieran la unión con el factor B que provoca su activación o con el factor H que cancela la progresión de la reacción. Otra proteína presente en el plasma, el factor D un factor realmente importante en algunas situaciones como se verá mas adelante, provoca el clivaje del factor B cuando está unido a C3b para formar así la molécula compleja C3bBb o C3 convertasa de la vía alternativa que inicia la activación de la vía común C5-C9.
Las seis proteínas C3, B, D, H, I y P, estas 5 últimas también denominados factores, juntas efectúan las funciones de iniciación, reconocimiento, activación y regulación de esta vía que resulta en la formación
de una unión C3/C5 convertasas.
La regulación de la vía clásica y la alternativa son absolutamente necesarias para evitar el desencadenamiento de una activación desordenada (tabla 4).
Intervienen en esta regulación tres proteínas, la primera es el inhibidor del C1 que frena la formación de C3b combinándose e inactivando a C1r y C1s. Esto previene la formación de C3 convertasa o complejo C4b2b en demasía, el segundo inhibidor es el factor H que frena la producción de C3b inhibiendo la unión del factor B a la membrana que tiene adherido como vimos el fragmento C3b, previniendo así la continuidad del clivaje de B a Bb y frenando la producción de C3bBb o C3 convertasa de la vía alternativa.
El último inhibidor es el factor I que inhibe la producción de C3b con un mecanismo de clivaje sobre esta misma molécula que lleva a la formación de C3c y C3d que son inactivos. Este factor I actúa solamente sobre membranas celulares que tienen el fragmento C3b adherido, casi siempre los eritrocitos.
La C3 convertasa inicial como vimos, está activándose constantemente en el plasma a tasas muy bajas bajo una regulación positiva efectuada por la properdina o factor P, cuya función en la vía alternativa es la de unirse a los fragmentos C3b pegados a las membranas celulares y estabilizar a las convertasas C3 y C5; esta misma regulación, pero en sentido negativo, es producida por los factores H e I 6.
Se han descrito una amplia variedad de activadores de esta vía, que abarcan desde ciertos polisacáridos como por ejemplo, el bacteriano (LPS), la levadura (zymosan), o vegetales como la inulina, los polisacáridos, hongos, bacterias, virus, células de mamíferos, y agregados de inmunoglobulinas, como por ejemplo, fragmentos Fab de IgA o IgE, aunque no se conoce todavía qué tienen en común estas estructuras que provocan el reconocimiento de esta vía de activación del complemento.
Todas las vías de activación contienen una enzima que cataliza la formación de C3 convertasa que a su vez genera C5 convertasa. Ésta provoca el clivaje de
la molécula de C5 y la formación del complejo de ataque a la membrana o MAC que está compuesto por cinco proteínas precursoras C5, C6, C7, C8 y C9.
La activación del MAC (C5-C9) es una consecuencia de la activación tanto de la vía clásica como la alternativa en la superficie de una célula. La convertasa C5 es generada por el clivaje de C5 por cual-quiera de las vías de activación que generan un pequeńo fragmento peptídico el C5a ( que es una potente anafilotoxina ) y un fragmento grande C5b.
Hasta este punto todas las reacciones son enzimáticas pero a partir de aquí las uniones ya no tienen este carácter y la unión de C5-C6 y C7 forman complejos no enzimáticos. El agregado de C7 precisamente transforma al complejo de hidrofílico en hidrofóbico, estado que hace a este complejo insertarse en la capa bilipídica de la membrana celular, momento en que se agregan C8 y 14 moléculas de C9 por cada complejo C5-C8 que terminan la formación del complejo MAC.
La molécula "asesina" es el MAC que constituye una organización supramolecular compuesta por más de veinte proteínas con un peso molecular de 1,7 millones Dalton. El MAC se une con la membrana celular de la célula "blanco" a través de un sitio de unión metaestable formándose una unión muy fuerte con la capa bilipídica de la misma.
Los eventos finales son el desdoblamiento, la oligomerización, y la polimerización de C9 (C9RP o proteína relacionada con C9) que provoca el debilitamiento de la estructura de la membrana y la formación de orificios y canales transmembranosos que llevan a la lisis osmótica de la célula.
Cuando la formación del complejo MAC llega a la situación de complejo C5b-8 cada una de estas moléculas actúa como receptor de un número
múltiple de moléculas C9 denominada "citolisina", "perforina", o "proteína formadora de poros", lo que parece facilitar la inserción de C9 en el núcleo hidrocarbonado de la membrana celular.
La unión del MAC es regulada por la proteína S7 (inhibidor primario del suero, cofactor del sistema de anticoagulación), que previene precisamente la polimerización de C9 y que está presente en el plasma, además de los factores de restricción homólogos que están presentes en la membrana celular. Este mecanismo de lisis celular por acción del complemento está presente en un gran número de células como los eritrocitos, las plaquetas, los linfocitos y en los virus con membrana lipoproteica.
La última de las vías de activación del complemento descripta es el de las "lectinas de unión con la manosa" o (MBP); fue en 19768 cuando al estudiar un grupo de nińos que tenían un retardo en el crecimiento y, al mismo tiempo padecían infecciones piógenas recurrentes, sus sueros carecían de la capacidad de opsonizar la levadura con C3 y que este defecto estaba relacionado con el nivel disminuido de las lectinas de unión con la manosa. Las "lectinas de unión con la manosa" son sustancias de la familia de las lectinas calcio dependientes denominadas "colectinas", y que resultaron ser homólogas en su estructura a la molécula de C1q del complemento.
Estas lectinas que son componentes normales del sistema inmunitario innato se unen a las membra-nas cuando están presentes los grupos terminales de la manosa de las membranas de una gran variedad de bacterias. Las lectinas de unión con la manosa se unen a unas serinoproteasas denominadas MASP-1, MASP-2 y MASP-310 en la superficie de las células bacterianas.
En el suero las MASP están presentes como proenzimas consistentes en una simple cadena de polipéptidos que se activan por clivaje. La MASP-1 activada cliva a C3 y a C2, mientras que la activación de MASP-2 cliva a C4 y C2. La función de MASP-3 no es conocida aún.
MASP-1 es capaz de clivar por si sola a la molécula de C3 e iniciar la activación, mientras que MASP-2 actúa en forma similar a C1s para llegar a la formación de complejos C4b2a que conforman a la enzima C3 convertasa de la vía clásica.
Las tres sendas convergen al punto de clivaje de la molécula central C3 donde se inicia el segmento común y que es precisamente la formación del complejo MAC o complejo de ataque a la membrana que provoca la lisis de la célula.
Resumiendo:
La vía clásica esta compuesta por la intervención de cuatro proteínas plasmáticas: C1, C2, C3, y C4.
La vía alternativa está constituida por pequeńas cantidades de C3b y proteínas conocidas como factores, tales como el factor B, factor D, incluyendo además a factores inhibidores como el factor H y el factor I (ver tabla 3), además de favorecedores como la Properdina o factor P.
La vía de las lectinas incluye la intervención de C2, C3, y C4 además de algunas de las lectinas calcio dependientes que son homólogas al componente C1 del complemento y que activan a las serinoproteasas MASP1 y MASP2.
El componente C3 es común a las tres vías.
El complejo de ataque a la membrana o MAC está compuesto por cinco proteínas C5, C6, C7, C8, y C9.
Un hecho fundamental. Las tres vías de activación convergen en un hecho común a ambas como es la generación de C3 convertasa, una enzima destinada a clivar a la molécula C3. Esta proteína es el centro de la cascada y contiene en su molécula un puente tioester.
El clivaje de C3 por la C3 convertasa da como resultado dos fragmentos proteicos C3b y C3a, en este preciso momento el puente tioester interno que queda en el fragmento C3b se activa por unos breves instantes permitiendo la unión de este fragmento a los grupos hidroxilos de los hidratos de carbono y de las proteínas de la vecindad. Luego de ese brevísimo instante toda molécula de C3 que no se una en este mecanismo es inactivada casi en forma inmediata por la unión con moléculas de agua.
La unión de C3 a los grupos carboxilos es el hecho clave de la cascada del complemento. Este fenómeno
permite marcar a los microorganismos como extra-ńos con la adhesión de las moléculas de C3b a la membrana (opsonización) .
Luego de este evento los fragmentos adheridos actúan como foco de activación de más complemento alrededor del microorganismo, lo que requiere la intervención del factor B o por otro lado la inclusión de la molécula del factor I que regula el catabolismo del fragmento C3b, provocando además la activación de anafilotoxinas (que tienen funciones quimiotácticas, activan a los fagocitos, inducen la degranulación de basófilos y células cebadas y producen contracción del músculo liso) y la formación del complejo de ataque a la membrana sobre la membrana del patógeno invasor.
Las Deficiencias del Complemento
Las deficiencias hereditarias del complemento provocan, por lo general, un aumento de la susceptibilidad para las infecciones piogénicas, tres son las deficiencias más importantes que muestran este defecto,
a. Deficiencia de la actividad opsónica - Aumento de la susceptibilidad a los organismos piógenos
Esta patología se observa en los pacientes que tienen defectos en la producción de anticuerpos, defectos en las proteínas componentes de la vía de activación clásica del complemento o defectos en la función fagocítica; estos trastornos suelen provocar aumento de la susceptibilidad a infecciones por Haemophilus influenzae y Streptococcus pneumoniae
b. Deficiencias de la actividad lítica - Aumento de la susceptibilidad a las infecciones por Neisseria
Para la defensa contra la Neisseria mengitidis es necesario que esté conservada la posibilidad de la formación del complejo de ataque a la membrana (MAC) debido a que el mecanismo de lisis es el de mayor prevalencia en la muerte de estos microorga-nismos que son en cambio capaces de sobrevivir intracelularmente.
c. Deficiencias en la función de la vía de las lectinas de unión con la manosa - Infecciones en nińos
En este grupo de nińos se apreció que la falla es especialmente importante en el período en que el sujeto pierde los anticuerpos maternos adquiridos pasivamente y el momento de la adquisición de los anticuerpos maduros propios11. La deficiencia se produce por una mutación génica en tres puntos de la lectina de unión con la manosa. Esta propensión a las infecciones recurrentes en los individuos afectados parece ser compensada con una ventaja en la vida adulta de estas personas que presentan una protección por ej. contra las infecciones por mico-bacterias12 y otras.
Al mismo tiempo que la cascada del complemento es un componente esencial de los mecanismos inmunes innatos y adquiridos, con la evolución de los organismos vivos algunos microorganismos logran utilizar precisamente los mecanismos de acción del complemento para penetrar en las células. Como ejemplo se puede citar que el virus de Epstein Barr utiliza
precisamente el receptor de tipo 2 del complemento de los linfocitos B, también conocido como CD21, como receptor de la proteína gp 350/220 de su cápsula13-14.
Otros organismos como el virus del HIV15 y las micobacterias16-17 activan al complemento del organismo huésped y provocan que la molécula C3b se una a la superficie de la célula lo que le permite al microorganismo penetrar en la misma a través del receptor del C3 del complemento.
También las bacterias pueden eludir la acción del complemento mediante el engrosamiento de sus cápsulas o la expresión en las mismas de proteínas que inhiban al complemento, por ejemplo el estrepto-coco A utiliza a la proteína M para unirse al factor H, que aumenta el catabolismo de C3b y reduce la formación de la C3 convertasa18 o aun inactivando al factor C5 por la secreción de una peptidasa que inactiva a los factores quemotácticos derivados del complemento como C5a19.
Los virus, como comenta Lubinsky20, disponen de tres mecanismos para evadir la acción del complemento:
1) Incorporan proteínas reguladoras del complemento en su envoltura como el herpesvirus, el virus vaccinia y el HIV.
2) Otros contienen proteínas que imitan a alguna de las proteínas reguladoras del complemento como la gC o glicoproteína C que es una proteína reguladora de la actividad de C3b, lo que le provee una ventaja de sobrevida al virus al inhibir funciones inmunes.
3) Una tercera posibilidad es que posean en su constitución una proteína de diferente estructura pero con funciones similares a las del complemento.
Otras deficiencias
Aunque excede el alcance de esta actualización debemos citar, por lo menos, que hay otras condi-ciones con deficiencias de factores del complemento:
a) Deficiencia del inhibidor del C1 del complemento que provoca la pérdida de la regulación de la actividad de C1 y falla en la actividad de la kalicreina provocando angioedema.
b) Deficiencia de CD59 que provoca fallas en la prevención de la formación del MAC en las células autólogas lo que lleva a la hemólisis y trombosis.
c) Deficiencias de C1q, C1r y C1s, C4 y C2 lo que provoca fallas en activación de la vía clásica y aparición de Lupus Eritematoso Sistémico.
d) Deficiencias de factor H e I provocando fallas en la regulación de la activación de C3 y deficiencia secundaria severa de esta fracción, provocando síndrome urémico hemolítico y glomerulonefritis membranoproliferativa.
Por otro lado la activación del complemento está evidenciada en un gran número de afecciones, algunas de ellas citadas en la tabla 5.
El factor nefrítico C3
El factor nefrítico C3 es un autoanticuerpo dirigido contra el factor B activado en la vía alternativa del complemento. Se lo encuentra presente comúnmente en el suero de los pacientes con glomerulonefritis membranosa proliferativa pero también está asociado con la lipodistrofia parcial o enfermedad de Barraquer - Simons21, un síndrome de rara ocurrencia (200 casos en la literatura mundial) casi siempre asociado a la glomerulonefritis post-estreptocócica, el LES o la glomerulonefritis idiopática rápidamente progresiva22.
En la glomerulonefritis membranoproliferativa existe una proliferación mesangial, un engrosamiento de la pared de los capilares y la existencia de depósitos subendoteliales de inmunoglobulinas y de C3.
La coexistencia de la lipodistrofia parcial (LP) es una condición desfigurante que coincide con este tipo de afecciones, caracterizada por una pérdida del tejido adiposo desde la cintura para arriba respetando a las piernas. La pérdida de grasa en este síndrome siempre fue considerada un misterio hasta que en 1989 fue aislada una substancia denominada adipsina23 en las células adiposas, con la característica de que comparte su secuencia de aminoácidos en un 98 % con el factor D del complemento así como su actividad enzimática.
La función del factor D, como vimos, es la de clivar al factor B cuando éste se encuentra unido al componente C3 facilitando la conformación y estabilización de la C3 convertasa (C3bBb). La particula-ridad de que la adipsina, símil al factor D, se
encuentra distribuida con un gradiente precisamente de mayor concentración en la mitad superior del cuerpo, explicaría la anormal distribución de la pérdida de grasa en estos cuadros clínicos, en los que la existencia del factor C3 nefrítico estabiliza a la C3 convertasa que se encuentra en la vecindad de los adipocitos.
Esta cantidad de enzima C3 convertasa estabilizada puede clivar suficiente cantidad de C3 con la formación de complejo de ataque a la membrana (MAC) en la vecindad de las células adiposas que de esta manera sufren la lisis.
El adipocito, es el mayor productor de factor D en el organismo aunque también está demostrado que producen factor C3 y factor B siendo activadores de la vía alternativa24 tanto in vitro como in vivo.
Cuando esta vía es desregulada por la presencia del factor nefrítico C3, se produce la destrucción de la célula grasa llevando a la expresión clínica de los raros casos de lipodistrofia parcial.
La explicación de la existencia de C3a en el adipocito, que se genera cuando C3 es clivado, es que C3a tiene además de su participación en la cascada del complemento, una potente actividad de proteína estimuladora de la acilación, mecanismo que promueve la esterificación de ácidos grasos en triglicéridos. Además C3a aumenta el transporte de la glucosa a través de la membrana del adipocito y por otro lado aumenta la actividad de la diacilglicerol aciltransferasa que tiene un efecto de marcado aumento de la síntesis de triglicéridos.
Como el papel fisiológico del adipocito es actuar como reserva de energía, acumula triglicéridos en tiempos de abundancia para su utilización ante la necesidad. La célula grasa expresa todos los componentes requeridos para la generación local de
C3a. El factor D aumenta con el ayuno o en estados catabólicos y disminuye su concentración en el adipocito en modelos de obesidad. Así, el adipocito puede modular su capacidad de activar la vía alternativa del complemento de acuerdo a las necesidades de liberación o acumulación de triglicéridos.
En la glomerulonefritis membranoproliferativa tipo II puede producirse un mecanismo similar desde que las células renales también expresan componentes del complemento y por aumento de la estabilidad de la C3 convertasa por la presencia del factor nefrítico C3 podrían producirse los depósitos densos subendoteliales de inmunoglobulinas y C3.
Al respecto cabe decir que las células renales expresan también componentes del complemento, especialmente C3 y C4 que son sintetizadas y secretadas por las células epiteliales de los túbulos renales, las células mesangiales y las epiteliales glomerulares. Esta expresión de componentes del complemento por el rińón es aumentada por la presencia de citoquinas proinflamatorias , por lo que la sobreactividad de la vía de activación alternativa por la presencia del factor nefrítico C3 tiene consecuencias nefastas para el rińón.

BIBLIOTECA
1- Bordet J . Sur le mode d'action des sérums préventifs. Ann Inst Pasteur 1896;10: 193-219.
2-Edward TG, Befus AD. The role of complement in the induction and regulation of immune responses. Immunology 1984; 51:207 .
3-Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC Studies of Group B Streptococcal Infection in Mice Deficient in Complement Component C3 or C4 Demonstrate an Essential Role for Complement in both Innate and Acquired Immunity. Proc. Natl. Acad. Sci. 1995; 92 (25): 11490-11494
4-Wurzner R, Orren A, Lachmann PJ. Inherited deficiencies of the terminal components of human complement. Immunodefic Rev 1992;3:123-47
5-Walport MJ. Complement New Engl J Med 2001;344: 1058-66, 1140-1144
6-Fearon, D, T; Austen, K, F. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase
J.Exp.Med. 1975;142: 856-863.
7-Preissner KT, Wassmuth R, Muller-Berghaus G. Physicochemical characterization of human S-protein and its function in the blood coagulation system. Biochem. J. 1985, 231 (349-355)
8-Soothill JF, Harvey BA. Defective opsonization: a common immunity deficiency. Arch Dis Child 1976;51:91-99.
9-Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet 1989;2:1236-9.
10-Matsushita, M.Complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J. Immunol. 2000; 164:2281-2284,
11-Turner MW, Super M, Singh S, Levinsky RJ. Molecular basis of opsonic defect in immunodeficient children. Clin Exp Allergy. 1991;21(suppl 1):182-8.
12-Bellamy R, Ruwende C, McAdam KP, et al. Mannose binding protein deficiency is not associated with malaria, hepatitis B carriage nor tuberculosis in Africans. QJM 1998;91:13-18.
13-Cohen, JI. Epstein-Barr Virus Infection New Engl J Med 2000, 343:481-492.
14-Fingeroth JD, Weis JJ, Tedder TF, Strominger JL, Biro PA, Fearson DT. Epstein-Barr virus receptor of human B lymphocytes in the C3d receptor CR2. Proc Natl Acad Sci U S A 1984, 81:4510-4.
15-Stoiber H, Clivio A, Dierich MP, Role of complement in HIV infection. Annual Review of Immunology 1997, 15: 649-674
16-Cywes C , Hoppe H C, Daffé M , Ehlers M R .Nonopsonic binding of Mycobacterium tuberculosis to complement receptor type 3 is mediated by capsular polysaccharides and is strain dependent. Infect Immun. 1997; 65 (10): 4258-4266
17-Schorey J S, Carroll M C., Brown E J. A Macrophage Invasion Mechanism of Pathogenic Mycobacteria Science 1997; 277: 1091-1093
18-Fischetti V A , Horstmann R D , Pancholi V Location of the complement factor H binding site on streptococcal M6 protein. Infect Immun. 1995 January; 63 (1): 149-153
19-Anderson ET, Wetherell MG, Winter LA, Olmsted SB, Cleary PP, Matsuka YV. Processing, stability, and kinetic parameters of C5a peptidase from Streptococcus pyogenes. Eur J Biochem. 2002 Oct;269(19):4839-51.
20-Lubinski J, Nagashunmugam T, Friedman HM . Viral interference with antibody and complement
Seminars in cell & delopmental biology, 1998;9,(3):329-337
21-L. Barraquer Roviralta: Histoire clinique d'un cas d'atrophie du tissue cellulo-adipeux. Barcelona, 1906.
22-Hartmut H.-J. Schmidt, Janine Genschel, Peter Baier, Martina Schmidt, Johann Ockenga, Uwe J. F. Tietge, Marcus Pröpsting, Carsten Büttner, Michael P. Manns, Herbert Lochs, and Georg Brabant
23-Dyslipemia in Familial Partial Lipodystrophy Caused by an R482W Mutation in the LMNA Gene J. Clin. Endocrinol. Metab. 2001; 86: 2289-2295
24-Rosen BS, Cook KS, Yaglon J, et al. Adipsin and complement factor D: an immune-related defect in obesity. Science 1989; 1483-1487.
25-ChoyI LN, Rosen BS, Spiegelman BM. Adipsin and an andogenous pathway of complement from adipose cells. J Biol Chem 1992; 267: 12736-12741.
26-Makrides SC. Therapeutic Inhibition of the Complement System. Pharmacol Rev.1998;50, (1), 59-88.
|