|
Tan antiguos como la vida los movimientos de entrada y salida de los iones tienen que ver con los comienzos de ella misma. Siguieron quiz�s hace unos 3.500 millones de a�os a la afectaci�n de la s�ntesis de productos g�nicos y al aparecer los primeros mecanismos regulatorios con la s�ntesis de genes alterados o quiz�s un poco m�s tarde hace unos 1.400 millones de a�os cuando los eucariotas necesitaron sistemas elaborados de membranas celulares que lograran controlar su composici�n intracelular (Fig
1) y que hicieron posible a trav�s de la evoluci�n la adaptaci�n a los cambios del medio ambiente para poder producir nuevos constituyentes y adquirir nuevas funciones. Aunque se hacen absolutamente necesarios hace unos 600 millones de a�os, en forma cierta, con la aparici�n de organismos multicelulares complejos que hizo imprescindible la existencia de un �rgano contr�ctil como el coraz�n (i�n dependiente) que fuera capaz de hacer llegar a todas y cada una de las c�lulas de los �rganos el ox�geno y los nutrientes as� como retirar los desechos que se fueran produciendo.
El ancestro primitivo m�s antiguo de los canales i�nicos, como hoy los conocemos, fue una mol�cula mucho m�s simple que las actuales y con un solo dominio algo similar al del canal de potasio; luego junto con la aparici�n de los eucariotas, este gen productor de las prote�nas del canal i�nico sufri� duplicaciones y divergencias que dieron lugar a la aparici�n del canal pot�sico tal como lo conocemos hoy, con sus dominios unidos en forma no-covalente y luego a la formaci�n de estructuras m�s complejas como el canal de calcio que finalmente por nuevas mutaciones di� lugar a la formaci�n de los canales de sodio que son capaces de generar potenciales de acci�n los que producen despolarizaci�n de las c�lulas m�s r�pida y eficientemente que los de calcio.
En los �ltimos a�os la medicina molecular fue obteniendo enormes avances en los conocimientos en este campo, muchos de los cuales est�n motivados por el adelanto espectacular en el entendimiento de la composici�n del genoma humano en lo que respecta a la caracterizaci�n de genes ligados a distintos s�ndromes cl�nicos. Un buen ejemplo de ello lo constituye el campo de la investigaci�n sobre la fisiopatolog�a de los canales i�nicos.
Hoy en d�a est�n descritas un n�mero considerable de enfermedades hereditarias de los canales i�nicos, conocidas en su conjunto como "canalopat�as".
En general la patolog�a de este sector se relaciona con los canales sensibles al voltaje y/o a los canales sensibles a la excitabilidad celular, siendo la activaci�n y la inactivaci�n de los canales de Na+ y K+ la base del potencial de acci�n que en definitiva es el responsable de permitir a las c�lulas excitables conducir la informaci�n, que controla a su vez una gran cantidad de acciones fisiol�gicas entre las que, solo por citar a las m�s relevantes est�n:
la propagaci�n del impulso nervioso y
la determinaci�n del ritmo card�aco.
As� consecuentemente muchas de estas enfermedades hereditarias por tras-tornos en la mutaci�n g�nica se traducen en des�rdenes neuromusculares y trastornos del ritmo card�aco.
Para comprender a las canalopat�as es necesario revisar la fisiolog�a de los canales i�nicos, �stos como dijimos est�n constituidos por t�neles conformados por prote�nas que atraviesan la membrana celular y donde se generan y orquestan miles de se�ales el�ctricas que pasan a trav�s del cerebro, el coraz�n y los m�sculos cada segundo durante toda la vida. As� los canales i�nicos se clasifican de acuerdo al tipo de i�n que permite pasar, ya sea sodio, potasio, calcio o cloro.
La mayor�a de las prote�nas de los canales i�nicos est�n compuestas por subunidades usualmente cuatro que son designadas como a y b reunidas alrededor del poro selectivo i�nico (Fig
2).
Cada subunidad contiene dominios repetitivos largos compuestos por segmentos con peque�os dominios a-h�lices que atraviesan la membrana celular (I,II,III y IV), cada uno de los cuales contienen seis unidades helicoidales transmembranosas (S1 - S6), en forma generalizada el segmento S4 tiene como funci�n el de activador o volt�metro, mientras que los segmentos S5 y S6 conforman el canal de paso de los iones a trav�s del poro, por �ltimo cada dominio est� conectado con el otro por medio de una cadena de uni�n las que a su vez tiene funciones especializadas de sensores el�ctricos actuando algunas como tapones de los extremos interiores de los canales, siendo las cadenas aminoterninales N las destinadas a la oclusi�n r�pida de la permeabilidad i�nica.
Figura 1-
Esquema de la evoluci�n de los canales i�nicos

Figura
2- Incrustada dentro de una membrana celular se esquematiza una
subunidad alfa de un canal i�nico con sus seis segmentos trasmembranosos.
S1 S6, que forman la estructura central de un canal de sodio, calcio o
potasio. En la cola aminoterminal existe una cadena con una
"bola" oclusiva que actua en forma r�pida para cerrar el
poro. los genes de un canal de sodio y calcio repiten esta
estructura seis veces mientras que los destinados a formar canales de
potasio solo lo hacen una vez. N Engl J. Med 1997,336:1578. (modif)

Los canales de sodio y calcio tienen sus dominios unidos en forma covalente, en cambio los canales de potasio no est�n unidos de esa manera. Adem�s los canales de sodio y calcio est�n codificados por genes que codifican cuatro repeticiones de la subunidad b�sica mientras que los espec�ficos de los canales pot�sicos solo lo hacen en una oportunidad (Fig
3), estando esos canales pot�sicos activados por voltaje Kv compuestos por una subunidad simple.
Un hecho fundamental en la fisiolog�a de los organismos vivos de todos los g�neros, desde los m�s rudimentarios seres unicelulares hasta aquellas c�lulas constituyentes de los complejos sistemas y tejidos que conforman el organismo de los seres humanos, es que siempre presentan una polaridad el�ctrica definida existiendo una diferencia de voltaje el�ctrico entre el interior citoplasm�tico y el medio extracelular que rodea a las c�lulas.
Esta diferencia de voltaje se desarrolla a trav�s de la membrana celular y se debe a dos hechos fundamentales
Figura 3.
Esquema de un canal pot�sico Kir, conductores eficientes de corriente
electrica hacia el interior celular, est� compuesto solo dos segmentos
transmembranosos M1 y M2.

1) la distribuci�n asim�trica de sustancias solubles con carga el�ctrica, los iones y
2) la permeabilidad selectiva de la membrana celular.
Esta propiedad de la permeabilidad selectiva es posible por la presencia de los canales i�nicos incrustados en la membrana, sustancias que son formadoras de poros submicrosc�picos. Los canales m�s importantes son los de Na+, Cl-, K+ y el Ca2+, cada uno de ellos tienen propiedades que permiten el paso del i�n espec�fico a trav�s de la membrana celular.
La magnitud de la diferencia de potencial que se produce por el flujo de los iones a trav�s de la membrana celular es del orden de apenas 10-12 amperes, de manera tal que los iones de Na+ o Ca2+ que est�n siempre en concentraci�n mayor en el espacio extracelular con respecto al intracelular, tienden a introducirse dentro de la c�lula cuando el poro espec�fico se abre, provocando la despolarizaci�n de la membrana. En cambio cuando los iones de K+ salen o los de Cl- entran, tambi�n a trav�s de los canales espec�ficos, el interior de la c�lula se negativiza hiperpolariz�ndose.
La cantidad de iones que son capaces de llegar a pasar por los poros i�nicos de una c�lula puede llegar a los 10.000.000 de iones por segundo1 .
Hace unos 50 a�os en un experimento cl�sico llevado a cabo por Hodgkin y Huxley2 demostraron la existencia de los potenciales de membrana midiendo la diferencia de voltaje entre el interior y el exterior de una neurona gigante de un calamar, fueron ellos mismos los que postularon la teor�a de que deb�an existir mecanismos de "apertura" y "cierre" el�ctricos relacionados con las necesidades fisiol�gicas, muy cercanas a las respuestas a los cambios de potenciales de membrana.
Se desprende de esto que los canales i�nicos pueden estar abiertos o cerrados seg�n convenga a la homeostasis y el proceso de pasaje de un estado al otro se denomina "gating" En este sentido tambi�n se conoce que existen tipos de canales i�nicos que pueden estar abiertos o cerrados en forma independiente del potencial de la membrana y en cambio otros canales i�nicos se abren o se cierran de acuerdo al estado de carga el�ctrica de la misma, diferenci�ndose a estos �ltimos que son los que interesan en patolog�a como:
1) canales i�nicos sensibles al voltaje de potencial de membrana,
2) canales i�nicos sensibles a est�mulos f�sicos o al calor o
3) canales i�nicos sensibles a interacciones con sustancias espec�ficas o ligandos.
Estos distintos tipos de canales tienen tres diferentes formas de estado, cerrados, abiertos o inactivos y usualmente responden a la despolarizaci�n de la membrana con una transici�n desde el estado cerrado hacia el abierto seguidos luego por una inactivaci�n intr�nseca3.
El pasaje de iones a trav�s de los poros no es una tarea exenta de gasto, de manera tal que un 30 % de la energ�a que consume una c�lula4 es utilizada en el mantenimiento de estos gradientes, la energ�a utilizada en esta acci�n es consumida por el canal en forma tal como lo hace un interruptor de contacto con una bater�a, de manera sumamente econ�mica y eficiente, con lo que solo se hace necesario para este enorme flujo de material la presencia de unos pocos miles de poros i�nicos de cada tipo en la membrana de una c�lula.
La activaci�n y la inactivaci�n de los canales de Na+ y K+ constituyen las bases de los potenciales de acci�n, que son las se�ales que como ya se cit�, permiten a las c�lulas excitables de los eucariotas conducir la informaci�n necesaria para cumplir con un amplio rango de eventos fisiol�gicos .
Los canales de Na+ y Ca2+ son los encargados de introducir corrientes despolarizantes dentro de las c�lulas, la transici�n del estado fisiol�gico de reposo cerrado al estado abierto de activaci�n ocurre simult�neamente a medida que los canales de Na+ y Ca2+ ven aumentadas las probabilidades de cerrarse debido a las se�ales inducidas por la misma corriente de despolarizaci�n.
Adem�s los canales de Ca2+ no solamente contribuyen a la polarizaci�n de la membrana, sino que tambi�n juegan un importante papel en el control de la entrada a la c�lula de este fundamental regulador intracelular. Para esto, una vez dentro del espacio intracelular el Ca2+ interact�a con enzimas, cofactores y otros segundos mensajeros que influyen sobre mecanismos tales como la contracci�n muscular, la exocitosis, la activaci�n enzim�tica y la expresi�n g�nica.
Canales de potasio.
Los canales de potasio son considerados los m�s primitivos dentro de la evoluci�n de la familia de los canales i�nicos voltaje-dependientes.
El �ltimo miembro identificado en la familia de los canales i�nicos luego del canal pot�sico tipo Kv conocido en su forma de subunidad simple ya citada, es el denominado Kir (por K potasio, i ingreso, r rectificador) , caracterizado por su particularidad de poder conducir corriente dentro de la c�lula en forma mucho m�s efectiva que para extraerla, a ra�z de permanecer abierto en los per�odos de estado, estos canales determinan la existencia de los potenciales de membrana en todas las c�lulas en reposo4, y act�an en forma de rectificador hacia adentro de la corriente, obteni�ndose ese efecto por la salida de iones potasio K+ (Fig
4).
Fig. 4 Los canales i�nicos y la excitabilidad el�ctrica. Los canales i�nicos sensibles al voltaje son responsables de la generaci�n de se�ales el�ctricas de autopropagaci�n llamados com�nmente potenciales de acci�n, en la ilustraci�n se pretende diagramar el curso de un est�mulo sobre una fibra muscular card�aca.
Un est�mulo excitador provoca una despolarizaci�n r�pida moment�nea y parcial cuando atraviesa un umbral de voltaje, provocando la apertura de los canales de Na+ (1) causando la despolarizaci�n de la membrana de la c�lula. Los canales de Na+ son entonces r�pidamente inactivados lo que permite que el K+ ingrese y devuelva al potencial de acci�n a un voltaje de meseta (2). Esta meseta es mantenida por el equilibrio entre las corrientes que fluyen hacia el exterior y hacia el interior a trav�s de los canales de K+ y Ca2+ respectivamente. La inactivaci�n progresiva de los canales de Ca2+ y la activaci�n creciente de los de K+ repolarizan la c�lula (3) llevando a la membrana a un potencial de reposo (4) que se mantiene merced a los canales de K+ rectificantes tipo
Kir.

Este subcap�tulo de los canales de potasio Kir sirve para mostrar todo lo que se puede esperar en el progreso de una mejor comprensi�n de la fisiolog�a y la aplicaci�n a la farmacolog�a de estos conocimientos.
Los canales Kir son conocidos desde hace m�s de medio siglo6, pero reci�n ahora se han podido clonar los genes que codifican las prote�nas que los componen, del an�lisis de esta composici�n surgi� la noci�n que por lo menos siete tipos de canales Kir diferentes se encuentran en los seres humanos, los mismos tienen funciones muy espec�ficas y tan loca-lizadas que sus alteraciones producen fallas en �rganos determinados
(tabla 1).
Valga decir p.ej que los canales Kir1.1 se expresan predo-minantemente en la rama ascendente del asa de Henle donde reciclan el K+ y median en la secreci�n neta del mismo en el nefr�n distal y es all� donde juegan su papel de mayor importancia en el mantenimiento de la homeostasis de este i�n, permitiendo un eficiente flujo de grandes cantidades de K+ hacia los t�bulos colectores del nefr�n distal7.
Pero ya se han clonado distintas variedades de esta subfamilia de canales Kir1 que se expresan en diferentes segmentos de los t�bulos renales lo que sugiere una funci�n distintiva en el control de la secreci�n de potasio hacia la orina en el ri��n.
Se ha detectado que la mutaci�n del gen ROMK, que es el que produce las prote�nas de los canales Kir1.0 expresadas en el t�bulo distal del ri��n dan lugar a la manifestaci�n del s�ndrome de Bartter, una enfermedad autos�mica recesiva caracterizada por hipokalemia, p�rdida de sal, alcalosis, hipercalciuria, hiperaldosteronismo y presi�n arterial normal, cuadro en el que se produce una disminuci�n del K+ en la luz del t�bulo, lo que impide la reabsorci�n del ClNa a trav�s de los intercambiadores de Na+/K+/2Cl- provocando una p�rdida de sal en ese segmento.
A su vez la llegada de Na+ a los t�bulos distales facilita la reabsorci�n de Na+ intercambiado por K+ e H+. Adem�s la disminuci�n de la absorci�n de Na+ en la rama gruesa ascendente produce disminuci�n de la reabsorci�n de Ca2+ con la consiguiente hipercalciuria, nefrolitiasis y osteoporosis8.
Los dem�s componentes de esta familia de canales pot�sicos Kir tienen al parecer funciones bien definidas, los Kir2.0 juegan un papel importante en el mantenimiento del potencial de reposo y la modulaci�n de los potenciales que forman las ondas de acci�n gobernando a la fase de reposo del potencial de acci�n de las fibras musculares card�acas balanceando el contenido de K+, aunque este tipo de canal tambi�n se expresa en el cerebro y en los m�sculos esquel�ticos.
La subfamilia Kir3.0 se expresa principalmente tambi�n en el cerebro y en el m�sculo card�aco, y en este aspecto los de tipo Kir3.1 se los encuentran muy particularmente en el atrio y uni�ndose a los canales de tipo Kir3.4 forman un receptor conocido como KAch que tiene un papel importante en la disminuci�n de la frecuencia card�aca durante la estimulaci�n vagal.
A su vez en el sistema nervioso central los mismos canales Kir3.0 est�n involucrados en la acci�n inhibitoria de GABA, acetilcolina, adenosina, somatostatina y los p�ptidos opioi-des.
Los canales Kir4.0 est�n fuertemente expresados en el ri��n, m�s espec�ficamente en la membrana laterobasal del epitelio del t�bulo distal lo que sugiere que pueden estar contribuyendo a suplir de K+ a la bomba de Na+-K+.
El grupo de canales Kir6.2 forman junto con el receptor de la sulfonilurea el canal responsable en el p�ncreas de la secreci�n de insulina mediada por la glucosa en las c�lulas b (canal pot�sico sensible a la adenosina-trifosfato); los canales pancre�ticos KATP normales se cierran en respuesta a la hiperglucemia, llevando a la membrana celular a la despolarizaci�n por el ingreso de Ca2+ y la liberaci�n de insulina, durante la hipoglucemia la apertura de esos mismos canales provoca la hiperpolarizaci�n de la membrana de la c�lula b impidiendo la liberaci�n de insulina.
Mutaciones en los genes que producen las prote�nas del canal Kir6.2 y la prote�na asociada SUR1 provocan la aparici�n de la Hipoglucemia hiperinsulin�mica persistente de la infancia tambi�n conocida como Nesidioblastosis o Hiperinsulinismo familiar, mutaci�n que lleva a la p�rdida de la habilidad para regular la liberaci�n de insulina en respuesta a los cambios de concentraci�n de la glucosa de la sangre.
Estos mismos canales Kir6.2 y los receptores SUR2A en el coraz�n forman otro canal KATP que median en los potenciales de acci�n acortados en los cardiomiocitos isqu�micos los que les confieren resistencia a la hipoxia, lo mismo ocurre con los canales Kir6.2 y los receptores SUR2B en las c�lulas del lecho vascular. Los canales KATP se convierten tambi�n en blancos de las neurohormonas y agentes farmacol�gicos como sulfonilureas y drogas de apertura de los canales pot�sicos.
Los canales Kir7.1 se expresan sobre todo en el cerebro, intestino y ri��n siendo los responsables del mantenimiento del potencial de reposo en las membranas de las c�lulas de estos tejidos.
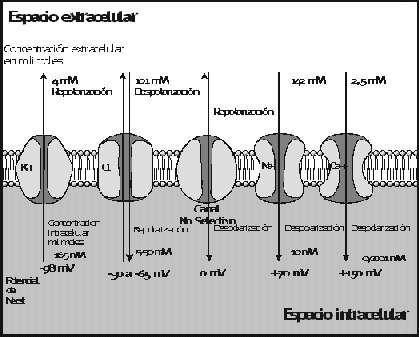
Figura 5 Esquematizaci�n de los distintos tipos de canales i�nicos, los diferentes gradientes intra y extracelulares y los resultantes potenciales de Nerst. Modificado de Ackerman; NEJM 336:1575-1586 modif.
Canales de sodio
Hablando de los seres humanos, �stos poseen por lo menos diez genes que codifican las unidades a de los canales de Na+ y la mayor�a de estos genes se expresan en los tejidos parti-cularmente excitables como el cerebro, los nervios perif�ricos y los m�sculos estriados. Las anomal�as de estos genes dan entonces lugar a diferentes formas patol�gicas como la par�lisis peri�dica hiperkal�mica, la paramioton�a cong�nita, la
mioton�a agravada por potasio, y tambi�n el s�ndrome del QT largo (tipo III).
Canales de calcio
En lo que respecta a los canales de Ca2+ existen ocho genes que codifican para seis variantes de canales de este i�n (tipos T, L, B, N, P y R), as� los canales de tipo L son sensibles a la acci�n de las dihidropiridinas (DHP) (nifedipina), fenilaki-laminas (verapamil) y las benzotiacepinas (diltiazem), por lo que se le ha adjudicado la denominaci�n err�nea de receptor de la dihidropiridina, (sugiriendo que este canal ser�a sensible a un ligando con una sustancia espec�fica cuando en realidad es voltaje dependiente).
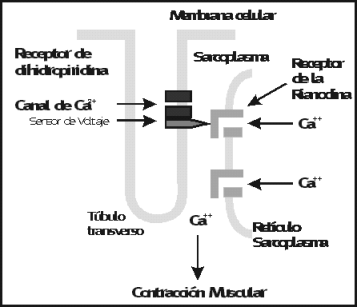
Otro tipo de canal de Ca2+ que interesa en cl�nica es el (RYR) o receptor de rianodina, as� denominado por su sensibilidad al alcaloide vegetal la Rianodina. Este canal posibilita la salida del calcio del ret�culo sarcopl�smico (RS) o el ret�culo endopl�smico (RE) , un paso esencial en la contracci�n del m�sculo esquel�tico (RYR1), y del m�sculo card�aco y los m�sculos lisos (RYR2). Los receptores de las dihidropiridinas (DHP) y de la rianodina (RYR1) est�n estrechamente relacionados con la contracci�n m�sculo esquel�tica.
Los receptores de las dihidropiridinas (DHP) est�n situados dentro de la membrana tubular transversa en las fibras musculares, durante la despolarizaci�n la activaci�n de los canales L lleva a cambios conformacionales de ciertas cadenas intracelulares, que subsecuentemente abren a los canales RYR1, lo que permite que el calcio ingrese al citoplasma desde el (RS) ret�culo sarcopl�smico. Esta se�al de transmisi�n entre el T-tubular y el (RS) de membrana es denominada cupla de contracci�n-excitaci�n
(Fig. 6).
Por otro lado las mutaciones de los genes que sintetizan las prote�nas del canal RYR1 causan la susceptibilidad para padecer hipertermia maligna y la enfermedad del n�cleo central (central core disease en ingl�s).
Figura 6.Esquema de cupla de contracci�n - excitaci�n muscular
Canales de cloro
Estos canales est�n presentes en la membrana plasm�tica de la mayor�a de las c�lulas involucradas en la regulaci�n del volumen celular, el transporte transepitelial, la secreci�n de fluidos de las gl�ndulas secretorias y la estabilizaci�n del potencial de membrana.
Los canales de cloro est�n subdivididos en tres grandes superfamilias.
1) Los receptores de glicina y GABAA (�cido gamma amino but�rico (GABA), identificada con el sub�ndice A (de all� su designaci�n como GABAA)). El �cido gamma amino but�rico es uno de los compuestos que transmiten informaci�n de una neurona a otra conocidos con el nombre gen�rico de neurotransmisores. Su acci�n sobre el receptor GABAA es el principal mecanismo de inhibici�n de la conducci�n del impulso nervioso en el sistema nervioso central. El receptor GABAA est� ubicado en la zona de la uni�n entre dos neuronas (la sinapsis) que median en el flujo de cloro dentro del sistema nervioso.
2)Los canales reguladores de la conductancia2 transmembranosa (CFTR) cuyos defectos dan lugar a la fibrosis qu�stica por alteraciones en pasaje del cloro a trav�s de las membranas. La conductancia es la medida de la facilidad con la que los iones fluyen a trav�s de un material y es expresado como la carga por segundo y por volt, distingui�ndose la conductancia de un solo canal y la del total de una c�lula o G.
3)Los canales de cloro activados por voltaje en las c�lulas excitables epiteliales.
A trav�s de estos distintos tipos de canales y sus variedades, muchas de las cuales seguramente se definir�n en un futuro, se mueven los iones forzados por las concentraciones relativas y sus gradientes tal como lo hacen en una soluci�n conectada a una bater�a. Existe en este caso un punto en el que las fuerzas qu�micas y el�ctricas est�n balanceadas, a este punto se lo llama potencial de Nerst. Aunque el concepto es mucho m�s complejo y excede el prop�sito de esta revisi�n una simplificaci�n del mismo dir�a que los potenciales de Nerst de los cuatro mayores canales i�nicos ser�an aproximadamente los siguientes (
Fig 5) :
Sodio + 40 mV
Potasio - 98 mV
Calcio + 150 mV
Cloro - 30 a - 65 mV,
siendo los signos positivos o negativos de los voltajes la expresi�n del potencial relativo intracelular con respecto a un electrodo de tierra. El potencial total de una membrana de una c�lula en un momento dado depende entonces de cuantos canales de cada i�n est�n abiertos o cerrados.
A partir de la observaci�n cl�nica y la investigaci�n de laboratorio muchas enfermedades hereditarias relacionadas con los canales i�nicos se fueron describiendo en las �ltimas d�cadas y se fue uniendo a la descripci�n cl�nica con la mutaci�n g�nica responsable de la misma, dando lugar a lo que se conoce actualmente como el cap�tulo de las canalopat�as, las mismas se tratan de enumerar en el siguiente cuadro, adjuntando al nombre de la afecci�n, el cromosoma en el que se localizan las mutaciones, el car�cter del defecto gen�tico y el tipo de canal i�nico afectado
(Tabla 2).
La Fibrosis Qu�stica
Se destaca entre todas las afecciones de los canales i�nicos asociadas a mutaciones hereditarias la fibrosis qu�stica, sobre todo porque uno de cada 22 individuos de raza blanca es portador de la mutaci�n del gen CFTR que codifica el canal de Cl- ubicado en el cromosoma 7q31. Una de cada 3000 personas de raza cauc�sica nace con las manifestaciones de la enfermedad, este gen mutado promueve un mal funcionamiento del canal de cloro de manera tal que impide el pasaje de este elemento a trav�s de la misma para su salida desde el medio intracelular. El defecto ubicado en el segmento apical de las c�lulas epiteliales impide asimismo el control de los canales de sodio que circula a trav�s de los canales ENaC aumentando la reabsorci�n de Na+ desde el l�quido extracelular.
La manifestaci�n cl�nica en estos pacientes es el aumento de la cantidad de Cl- en el sudor > 60 mmol/litro , poliposis nasal y enfermedad pulmonar, bronquiectasias, neumot�rax, cor pulmonale, obstrucci�n de las v�as biliares, la acumulaci�n de mucus que se asocia con insuficiencia pancre�tica, �leo meconial en el intestino del reci�n nacido, infertilidad por ausencia cong�nita de los vasos deferentes (azoospermia).
El 70 % de los portadores tiene una deleci�n de una fenilalanina en la posici�n 508 de la prote�na que conforma el canal de cloro, pero a�n as� esta mutaci�n permite cuando est� incorporada a la membrana celular el transporte de cloro; lo que en realidad sucede es que debido a un plegamiento conformacional an�malo24 esta prote�na se acumula en las organelas intracelulares y no llega a incorporarse a la membrana celular, lo que abona para la idea de que mediante una "chaperonina" que acompa�e el viaje de esta prote�na hasta la membrana podr� solucionarse en el futuro un porcentaje de los casos.
El 90 % de las muertes por esta afecci�n se producen por enfermedad pulmonar, el conocimiento del defecto en el canal i�nico ha llevado a intentar con suerte diversa el reemplazo del gen defectuoso mediante transferencia por portadores virales del gen normal25.
Los s�ndromes del QT largo.
Un segundo grupo de las canalopat�as est�n compuestas por aquellas que se manifiestan por un intervalo Q-T prolongado en el electrocardiograma (LQT), afecciones que se agrupan bajo el nombre de s�ndromes del QT largo y corresponden a las primeras arritmias de origen gen�tico descriptas que afectan a los potenciales de acci�n.
Desde un s�ncope inexplicado hasta la muerte s�bita, o el hallazgo asintom�tico de un ECG patol�gico, prolongaci�n del QT y anormalidades de las ondas T y U26 o muerte s�bita, 75 % de los casos son descubiertos por ex�menes de rutina los que pueden prevenir las graves manifestaciones cl�nicas de esta canalopat�a que en �ltima instancia es provocada por el aumento de la duraci�n de los potenciales de acci�n de las fibras musculares card�acas.
25 % de los casos son diagnosticados por ex�menes a consecuencia de un s�ncope inexplicado sin alteraciones de la salud previas que suceden a menudo durante el ejercicio o durante un estr�s emocional.
Estos s�ndromes cong�nitos de LQT tienen una incidencia bastante importante llegando a 1 de cada 10.000 personas.
Las mutaciones que provocan LQT han sido identificados en tres genes y �ltimamente un cuarto gen, cada uno codificando un canal i�nico27
(Fig. 7).
Figura 7- Ubicaci�n cromos�mica de los genes responsables de LQT. Ideogramas de los cromosomas 11, 7, 3, y 4 mostrando las localizaciones de los genes LQT1 LQT2 LQT3 Y LQT4. Roden D. Y col. Circulation. 1996;94:1996-2012.)
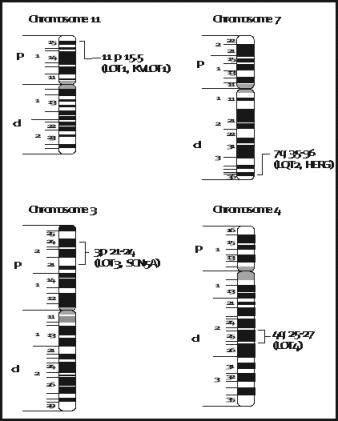
1) Entre los individuos afectados con LQT se han identificado mutaciones en KvLQT1 en el cromosoma 11 en familias portadoras de un gen recientemente clonado que codifica las prote�nas de un canal de potasio. El primer locus cromos�mico individualizado fue precisamente el ubicado en la regi�n telom�rica del cromosoma 11 (11p15.5) dando lugar al s�ndrome LQT1.
2) Mutaciones en el gen humano eter - a - go - go (HERG) que codifica el canal pot�sico, provoca la aparici�n de la enfermedad en familias portadoras de esta anomal�a en el cromosoma 7. En 1994, Jiang y col publicaron la evidencia de la ubicaci�n en el cromosoma 7q35-36 (causante del LQT2) en nueve familias.
3)En familias que son portadoras de mutaciones en el gen SCN5A en el cromosoma 3 ( 3p21-24) provoca la aparici�n del s�ndrome LQT3, este gen codifica el canal de sodio card�aco humano .
4)Ha sido identificado un cuarto sitio en el cromosoma 4 como 4q25-27 (LQT4 29), esta forma es rara porque hay una incidencia alta de fibrilaci�n auricular junto con el fenotipo de LQT, aunque se ha citado la existencia de un gen alterado que codifica para calcio-modulina quinasa.
Dos formas cl�nicas se han definido, una asociada a sordera cong�nita con caracter�sticas autos�micas recesivas denominado s�ndrome de Jervell y Lange-Nielsen30 , y la otra forma es la autos�mica dominante o s�ndrome de Romano-Ward 31 que es m�s com�n que el anterior y de frecuencia creciente en el tiempo seg�n los cardi�logos.
Las enfermedades miot�nicas
Por �ltimo aunque s�lo lo hacemos con un criterio de simplificaci�n, podremos citar a las mioton�as que comprenden a un grupo de afecciones que comparten el hecho de ser miot�nicas: relajaci�n muscular retardada despu�s de una contracci�n voluntaria o una estimulaci�n mec�nica.
Ellas son
1)La par�lisis hiperkal�mica peri�dica, episodios de par�lisis pasajera, enfermedad autos�mica dominante con accesos frecuentes, breves y provocados por el reposo post-ejercicio, el estr�s y/o la ingesti�n de ciertos alimentos y potasio. Son debidas a un defecto en los canales de Na+.
2)La paramioton�a cong�nita, consiste en una mioton�a loca-lizada acompa�ada de debilidad muscular, es prolongada e inducida por el fr�o. Se agrava con la actividad muscular, (mioton�a paradojal) en contraste con el mejoramiento de los s�ntomas observados con el ejercicio en los pacientes con mioton�as cl�sicas. Es debida a alteraciones de los canales de Na+.
3)La mioton�a cong�nita, en estos pacientes la contractura se resuelve con el ejercicio. La variante autos�mica dominante corresponde a la enfermedad de Thomsen. El "ejercicio isom�trico" que estos pacientes sobrellevan les da el aspecto de "gimnasta de circo". Se deben a alteraciones en los canales de Cl- y su forma autos�mica recesiva corresponde a la enfermedad de Becker.
4)Un cuarto grupo que incluir�amos en este apartado es el s�ndrome hipermetab�lico maligno o hipertermia maligna, que no es una enfermedad en el sentido estricto sino una predisposici�n gen�tica de algunos individuos a responder en forma anormal cuando son expuestos a anest�sicos vol�tiles o a relajantes musculares despolarizantes
(Fig. 6).
Es un cuadro inducido por drogas, potencialmente letal en los portadores de la anomal�a del canal de Ca2+. En estos casos una gran concentraci�n miopl�smica de Ca2+ sigue a la exposici�n del agente activador relajantes musculares despolarizantes, (succinidilcolina), todos los agentes anest�sicos vol�tiles (halotano, enflurano, isoflurano, sevoflurano y desflurano), cafe�na y todos los materiales de contraste para rayos X halogenados, adem�s influyen las situaciones de estr�s como la cirug�a, el embarazo, las infecciones y el estr�s psicol�gico), causando un aumento del metabolismo muscular con producci�n de calor que resultan en el tan temido s�ndrome de rigidez muscular, hipertermia, acidosis metab�lica, hiperkalemia e hipoxia.
Desde el punto de vista fisiol�gico la despolarizaci�n de la fibra muscular comienza en el final de la placa motora y se transmite a lo largo de la fibra muscular. El potencial de acci�n es transmitido a todas las fibrillas en la fibra muscular por el sistema tubular T los que luego:
sActivan la liberaci�n de Ca2+ desde las cisternas terminales (sacos laterales del ret�culo sarcoplasma).
sCa2+ se une a la Troponina
sEsto debilita la uni�n de la Troponina I con la actina
sPermitiendo a la tropomiosina moverse lateralmente, descubriendo el sitio de encaje para la miosina
sLa ATPasa de la miosina comienza la liberaci�n de energ�a del ATP
sLa miosina se adhiere a la actina
sSe produce un acortamiento progresivo del m�sculo
sSe produce el transporte de Ca2+ hacia dentro del ret�culo sarcoplasma
sFinalmente se produce la relajaci�n de las fibrillas musculares.
Desde el punto de vista terap�utico se utiliza el dantrolene, un inhibidor de la liberaci�n del Ca2+ del ret�culo sarcoplasma de la fibra muscular para intentar abortar la crisis.
La predisposici�n a la hipertermia maligna es gen�ticamente heterog�nea, y el responsable de la alteraci�n es un gen codificador del receptor de la rianodina en el m�sculo esquel�tico .
Por �ltimo cabe consignar a una forma al�lica de la hipertemia maligna que corresponde a la enfermedad muscular central o (central core disease), miopat�a autos�mica dominante con alteraciones estructurales en algunos tipos de fibras musculares. Estos pacientes son hipot�nicos en el nacimiento pero la fuerza muscular mejora con el tiempo.
El estudio y la descripci�n de las alteraciones del funcionamiento de los canales i�nicos no solo ayudar� a la comprensi�n de los pacientes de las afecciones antecitadas sino que tambi�n beneficiar� a la mejor elucidaci�n de numerosos cuadros de la cl�nica general.
Aunque existen numerosas publicaciones sobre el tema, se recomienda para una ubicaci�n actualizada de todas las alteraciones descriptas en forma muy detallada la p�gina de la Washington University School of Medicine, St. Louis, MO; Neuromuscular disease center
en http://www.neuro.wustl.edu/neuromuscular/index.html
1-Vega Hernandez A, Felix R. Fisiopatolog�a de los canales i�nicos sensibles al voltaje. Avance y perspectiva. 2001, 20: 83-96
2-Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952;117:500-544.
3-Armstrong CM, Hille B. Voltage-gated ion channels and electrical excitability. Neuron 1998;20:371-80
4-Ackerman MJ, Clapham DE.
Ion Channels - Basic Science and Clinical Disease. N Engl J Med. 1997; 336:1575-1586.
5-Jan LY, Jan YN. Potassium channels and their evolving gates. Nature 1994; 371:119-122.
6-Katz, B. Les constantes electriques de la membrane du muscle. 1949Arch. Sci. Physiol. 2, 285-299.
7-Wang, W., Hebert, S. C., and Giebisch, G. Renal K channels: structure and function. 1997 Annu. Rev. Physiol. 59, 413- 436
8-Simon, D. B., Karet, F. E., Rodriguez-Soriano,J., Hamdan, J. H., DiPietro, A., Trachtman, H., Sanjad, S. A., and Lifton, R. P. (1996) Genetic heterogeneity of Bartter's syndrome revealed by mutations in the Kchannel, ROMK. Nat. Genet. 14, 152-156
9-Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med 1997;336:1562-1567.
10-Davis PB, Drumm M, Konstan MW. Cystic fibrosis. Am J Respir Crit Care Med 1996 154:1229-1256.
11-Shimkets RA, Warnock DG, Bositis CM, et al. Liddle's syndrome: heritable human hypertension caused by mutations in the subunit of the epithelial sodium channel. Cell 1994; 79:407-414.
12-Malignat hipermetabolic disease en http://www.anaesthetist.com/index.htm accedido el 28-05-2002.
13-Aynsley-Green, A., Polak, J. M., Bloom, S. R., Gough, M. H., Keeling, J., Ashcroft, S. J., Turner, R. C., and Baum, J. D. Nesidioblastosis of the pancreas: definition of the syndrome andthe management of the severe neonatal hyperinsulinaemic hypoglycaemia. 1981; Arch. Dis. Child. 56, 496-508.
14-Karolyi, L., Koch, M. C., Grzeschik, K. H., and Seyberth, H. W. The molecular genetic approach to Bartter's syndrome. 1998 J. Mol. Med. 76, 317-325
15-Audesirk G, Armstrong D, van den Maagdenberg A, Atchison W, Shafer T, Fletcher C. Calcium Channels: Critical Targets of Toxicants and Diseases Environmental Health Perspectives Volume 108, Number 12, December 2000.
16-Brown KA, Al-Gazali LI, Moynihan LM: Genetic heterogeneity in Schwartz-Jampel syndrome: two families with neonatal Schwartz-Jampel syndrome do not map to human chromosome 1p34- p36.1. J Med Genet 1997 Aug; 34(8): 685-7 .
17-Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA,Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136-40.
18-Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1 A 4. Cell. 1996; 87 (3) :543-552.
19-Kraus RL, Sinnegger MJ, Glossmann H, Hering S, Striessnig J: Familial hemiplegic migraine mutations change alpha1A Ca 2+ channel kinetics. J Biol Chem 1998, 273:5586-5590.
20-Beck C, Moulard B, Steinlein O, Guipponi M, Vallee L, Montpied P, Baldy-Moulnier M, Malafosse A . A nonsense mutation in the alpha4 subunit of the nicotinic acetylcholine receptor (CHRNA4) cosegregates with 20q-linked benign neonatal familial convulsions (EBNI) Neurobiol Dis 1994; 1(1-2):95-9.
21-Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med. 1992;326:1108-1114.
22-Lennon VA, Kryzer TJ, Griesmann GE, O'Suilleabhain PE, Windebank AJ, Woppmann A, Miljanich GP, Lambert EH: Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med 1995, 332:1467-1474.
23-Scheinman SJ. Metabolic Causes of Tubulointerstitial Disease en: http://cnserver0 .nkf.med.ualberta.ca/cn/Schrier/Volume2/chapt11/ADK2_11_1-3.pdf accedido el 27-05-2002.
24-Maino R. El enfisema, la cirrosis, la trombosis, la demencia y las enfermedades conformacionales. Medicina Interna 2002;3(1): 3-10.
25-Alton EW, Geddes DM. Gene therapy for cystic fibrosis: a clinical perspective. Gene Ther 1995;2:88-95.
26-Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, Schwartz PJ, Towbin JA, Vincent GM, Lehmann MH, Keating MT, MacCluer JW, Timothy KW. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation. 1995; 92:2929-2934.
27-Roden D M, Lazzara R, Rosen M, y col. Multiple Mechanisms in the Long-QT Syndrome. Circulation. 1996;94:1996-2012.
28-Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995;81:299-307.
29-Schott J-J, Charpentier F, Peltier S, et al. Mapping of a gene for long QT syndrome to chromosome 4q25-27. Am J Hum Genet 1995;57:1114-1122.
30-Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J. 1957;54:59-68.
31-Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare in eta pediatrica. Clin Pediatr. 1963;45:656-683.
32-Ogawa Y. Role of ryanodine receptors. Crit Rev Biochem Mol Biol 1994;29:229-274.
|