|
|
SOCIEDAD DE MEDICINA INTERNA DE BUENOS AIRES |
|
|
SOCIEDAD DE MEDICINA INTERNA DE BUENOS AIRES |
|
Revista de la Sociedad
de Medicina Interna Localización Inusual de Linfoma
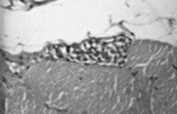
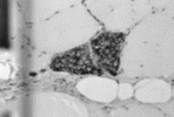
angiotrópico de células grandes El linfoma angiotrópico de células grandes (LACG) es una rara entidad gnosológica descripta por Pfleger y Tappeiner en 1959. Es una extrańa enfermedad caracterizada por la proliferación intravascular de linfocitos sin la presencia de células neoplásicas en los tejidos linfoides ni en sangre periférica. Por este motivo, esta entidad ha sido reportada bajo diferentes nombres como, por ejemplo, angioendoteliomatosis, linfomatosis maligna intravascular, angioendoteliomatosis sistémica, endotelioma, hemangioendoteliosis, entre otros. Todos estos términos sugieren un origen endotelial de la neoplasia. No mucho tiempo atrás, también se sugirió que esta enfermedad representaba la diseminación endovascular de carcinomas ocultos. La interpretación de la patología que nos ocupa como un infrecuente desorden proliferativo del endotelio endovascular fue hecha sobre la base de las evidencias de la microscopía óptica y electrónica así como por la determinación del antígeno relacionado con el factor VIII. De acuerdo con la evolución clínica, se solía distinguir entre formas benignas y malignas. Las variantes benignas se asociaban frecuentemente con endocarditis bacteriana con buena respuesta a los antibióticos y a los esteroides. Por su parte, las variantes malignas eran presentadas como fatales en pocos meses con o sin tratamiento, aunque se describieron algunos casos raros de remisión. Sin embargo, estudios recientes muestran claramente la naturaleza linfoide de esta patología y el término “linfoma angiotrópico de células grandes” se propuso como la designación más adecuada. El propósito de este artículo es comunicar a la comunidad científica un caso de LACG con localización sumamente infrecuente cual es el músculo esquelético en ausencia de compromiso sistémico. Preparado de la Pieza Anatómica
Caso clínico:
Discusión: Como vimos en la introducción (vide supra), estudios recientes muestran la naturaleza linfoide de esta entidad. El origen leucocitario se evidencia por la fuerte expresión del antígeno común leucocitario y las reacciones negativas para el antígeno relacionado con el factor VIII, excepto en algunas células atrapadas con las plaquetas y el trombo de fibrina (1). La proliferación neoplásica se produce dentro de la luz de los capilares y pequeńas venas y arterias, sin compromiso o afectación del tejido parenquimatoso perivascular. Los estudios inmunohistoquímicos demuestran claramente que estos tumores son, en su mayoría, originarios de las células B, aunque existen algunos casos correspondientes al linaje de linfocitos T. El origen de esta neoplasia es todavía desconocido; podría corresponder a una diseminación secundaria de una neoplasia linfoide oculta o bien a una lesión linfoide maligna intravascular primaria. La afinidad de las células tumorales por el endotelio capilar se relacionaría con los receptores linfoides para los antígenos de la membrana endotelial, como aquellos expresados en el sector postcapilar y venular. Se ha postulado que los linfocitos maduros expresan en su superficie receptores para las células endoteliales y que se unen a ellas, mientras que los precursores inmaduros se unen muy poco o nada. Entonces, las células neoplásicas podrían cambiar la expresión de sus receptores de superficie en el espacio intravascular y proliferar allí sin invasión parenquimatosa. Los linfocitos neoplásicos invaden los nódulos linfáticos de la misma manera que los linfocitos normales. Por lo tanto, la restricción de este linfoma al espacio intravascular podría deberse a alteraciones en el antígeno para el receptor Hermes3, un receptor para linfocitos ubicado en el endotelio venular. Una pobre expresión de este antígeno en las células linfomatosas impediría la adherencia y posterior invasión parenquimatosa de la neoplasia. (2) Sin embargo, algunos autores sugieren que el mecanismo descripto no sería el responsable de la localización endovascular y postulan que otro tipo de alteraciones moleculares, ya sea en la membrana de los linfocitos, ya sea en el endotelio de vasos de ciertos órganos, serían las responsables de este curioso fenómeno. (3) Aunque el tumor permanece clásicamente dentro de los vasos, se ha reportado un caso en el que el análisis citogenético de los mononucleares de sangre periférica ha revelado la presencia de células malignas en sangre circulante; estas células presentaban anormalidades en el cariotipo similares a aquellas encontradas en linfomas y leucemias. El análisis citogenético mostró dos clones relacionados anormales con cariotipo hiperdiploide y varias alteraciones cromosómicas comúnmente observadas en los desórdenes linfoproliferativos malignos (trisomía 7, trisomía 12, trisomía 18 y deleción del brazo largo q del cromosoma 6, etc.) así como una traslocación comprometiendo el brazo corto (p) de los cromosomas 1 y 3. Los autores concluyen que son necesarios nuevos estudios citogenéticos adicionales a fin de determinar si existe una alteración cromosómica estructural o numérica.(4) La enfermedad generalmente se disemina con rapidez y su curso es rápidamente evolutivo a pesar de la quimioterapia. Muchas veces el diagnóstico es postmorten. La mortalidad promedio es superior al 80% y el tiempo promedio de sobrevida es de unos 13 meses. (5) Las localizaciones más comunes son la piel y el SNC. No obstante, otros tejidos y órganos pueden verse afectados como, por ejemplo: rińón (6), próstata, hueso, páncreas, pulmón (7), corazón y suprarrenales. La mitad de los casos reportados presenta compromiso cutáneo caracterizado por hiperpigmentación, placas hipervascularizadas o nódulos. Otros hallazgos comunes en piel son telangiectasias, hemorragias y ulceraciones, e infiltrado dérmico inflamatorio adherido a los tejidos subyacentes. Los sitios predilectos son el tronco y las extremidades. Debe establecerse el diagnóstico diferencial con el eritema nodoso, la micosis fungoide, la paniculitis, la vasculitis granulomatosa, la sarcoidosis y el sarcoma de Kaposi. Casi la mitad de los casos también muestra compromiso neurológico caracterizado por accidentes cerebrovasculares y encefalopatía progresiva. Otros signos son: visión borrosa, episodios afásicos transitorios, obnubilación, debilidad, disminución o pérdida de la sensibilidad. La electroencefalografía y la tomografía axial computarizada pueden mostrar alteraciones significativas. Por el contrario, la angiografía suele ser normal. En síntesis, la literatura seńala que aproximadamente el 80% de los pacientes presenta signos o síntomas de compromiso cutáneo o del SNC. (8) A diferencia de las leucemias, la médula ósea no se afecta y la linfadenopatía es excepcional. (9) Los casos de compromiso del músculo esquelético son extremadamente raros. Glass et al han estudiado 114 casos publicados de LACG. Hallaron que la localización esquelética era sólo del 3% y siempre coexistía con localización neurológica. (10) Stroup et al (11) han descripto el caso de un varón de 68 ańos de edad quien presentó anorexia, pérdida de peso, debilidad y dolor lumbar. El paciente tenía, además, anemia, leucopenia y el recuento de plaquetas era de 93.000/mm. La biopsia de la médula ósea mostró un 65 a 79% de celularidad. Debido a la leucopenia se practicó una esplenectomía sin poder hallarse células neoplásicas. A causa de la debilidad muscular se realizó una biopsia muscular, la que permitió el diagnóstico de LACG. En este caso, los hallazgos histológicos fueron: numerosas dilataciones vasculares de diferente tamańo, con oclusión parcial o total de la luz por proliferación neoplásica de mononucleares; células neoplásicas grandes con núcleo hipercromático, de contornos irregulares y nucléolo prominente; las células endoteliales no mostraron atipía. Los estudios inmunohistoquímicos revelaron fuerte expresión del antígeno común leucocitario y las pruebas para keratina y antígeno relacionado con factor VIII fueron negativas; también revelaron expresión fenotípica de células B . Este caso que transcribimos es el único que hallamos en la literatura con compromiso de músculo esquelético y sin afección de piel y/o SNC. El caso que comunicamos en este artículo difiere del descripto anteriormente en que no presentaba compromiso sistémico de ningún tipo siendo la única manifestación la tumoración muscular biopsiada. Tanto la microscopía óptica, cuanto los estudios inmunohistoquímicos que llevamos a cabo, confirmaron que se trataba de un LACG correspondiente al linaje de células B. Es de destacar que, hasta el momento (más de 14 meses de realizado el diagnóstico) y a pesar de lo establecido en la literatura, la evolución del paciente es favorable con buena respuesta a la quimioterapia. Referencias Sepp, N et al. Intravascular lymphomatosis (angioendotheliomatosis). Evidence for a T-cell origin in two cases. Human Pathology, October 1990, vol.21, n ş 10, 1051 - 1058. 2 Yousem, SA; Colby, TV. Intravascular lymphomatosis presenting in the lung. Cancer, January 1990, vol. 65, 349 - 353. 3 Demirer, T; Dail, DH; Aboulafia, DM. Four varied cases of intravascular lymphomatosis and a literature review. Cancer, March 15, 1994, vol. 73, n ş 6, 1738 - 1745. 4 Molina, A et al. Immunohistochemical and cytogenetic studies indicate that malignant angioendotheliomatosis is a primary intravascular (angiotropic) lymphoma. Cancer, August 1990, vol. 66, 474 - 479. 5 Sheinani, K et al. Further evidence that “malignant angioendotheliomatosis” is an angiotropic large-cell lymphoma. New Engl J, 1986, vol. 314, n ş 15, 943 - 947. 6 D´Agati, V et al. Angiotropic large cell lymphoma (Intravascular malignant lymphomatosis) of the kidney: presentation as minimal change disease. Human Pathology, March 1989, vol. 20, n ş 3, 263 - 268. 7 Snyder, LS; Harmon, KR; Estensen, RD. Intravascular lymphomatosis (malignant angioendotheliomatosis) presenting as pulmonary hypertension. Chest, 1989, vol. 96, n ş 5, 1199 - 1200. 8 Levin, KH; Lutz, G. Angiotropic large-cell lymphoma with peripheral nerve and skeletal muscle involvement: early diagnosis and treatment. Neurology 47, October 1996, 1009 - 1011. 9 Carter, DK et al. Angiotropic large cell lymphoma (intravascular lymphomatosis) occurring after follicular small cleaved cell lymphoma. Mayo Clin Proc, September 1996, vol. 71, 869 - 873. 10 Glass, J; Hochberg, FH; Miller, DC. Intravascular lymphomatosis. A systemic disease with neurologic manifestations. Cancer, May 1993, vol. 71, n ş 19, 3156 - 3164. 11 Stroup, RM et al. Angiotropic (intravascular) large cell lymphoma. Cancer, October 15, 1990, vol. 66, 1781 - 1788. |
![]()